


参考资料
1.Best practices for single-cell analysis across modalities.Nature _论文_ & _大纲格式的介绍_
2.Luecken et al. (2022). Benchmarking Atlas for Single-Cell Analysis Tools. Nature Methods. _论文_
3.Stuart & Butler et al. (2019). Comprehensive Integration of Single-Cell Data. Cell. _论文_
4.scanpy tutorial held at Helmholtz Munich in July 2020 _Youtube视频_
5.组学之心 _知乎_ & _微信公众号_
6.小红书笔记 _写一个生信小白也能看懂单细胞测序分析_ & _挑战30天学习单细胞测序分析_
一、定义
测序 Sequncing
测序是破译 DNA 核苷酸顺序的过程,主要用于揭示特定 DNA 片段、完整基因组甚至复杂微生物组所携带的遗传信息。
但DNA测序无法完全揭示细胞的动态和实时操作。RNA 测序(RNA-Seq)通过检测基因表达谱,帮助研究细胞状态变化、基因亚型、基因融合等。RNA-Seq 不受先验知识限制,可捕获已知和新特征。RNA-Seq 主要遵循 DNA 测序方案,但包括逆转录步骤,从 RNA 模板合成 cDNA。
RNA 测序主要通过两种方式进行:对来自目标来源的混合 RNA 进行跨细胞测序(批量测序)或单独对细胞的转录组进行测序(单细胞测序)。在大多数情况下,混合所有细胞的 RNA 比实验复杂的单细胞测序更便宜、更容易。大量 RNA-Seq 会产生细胞平均表达谱,这通常更容易分析,但也隐藏了一些复杂性,例如细胞表达谱的异质性,这可能有助于回答感兴趣的问题。
测序简史
第一代测序 Sanger
- 背景:
- 1869 年,Friedrich Miescher 首次分离 DNA。
- 1953 年,Watson、Crick 和 Franklin 发现 DNA 的结构。
- 1965 年,Robert Holley 完成第一个 tRNA 测序。
- 1972 年,Walter Fiers 首次完成完整基因(噬菌体 MS2 外壳蛋白基因)测序。
- Sanger 测序:
- 由 Friedrich Sanger 开发,使用“链终止法: 通过在DNA合成过程中引入双脱氧核苷酸(ddNTP)随机终止链延伸,生成不同长度的片段以实现序列读取”(也称 Sanger 测序)。
- 1987 年,Leroy Hood 和 Michael Hunkapiller 开发 ABI 370,实现 Sanger 测序自动化,引入荧光染料标记。
- 优势:
- 简单且经济实惠。
- 错误率极低(<0.001%)。
- 局限性:
- 只能测序短片段 DNA(约 300-1000 bp)。
- 前 15-40 个碱基质量较差(引物结合区)。
- 700-900 个碱基后测序质量下降。
- 测序成本高于第二代和第三代测序。
第二代测序 Pyrosequencing
- 背景:
- 1996 年,Mostafa Ronaghi 等人引入焦磷酸测序技术,开启第二代测序时代。
- 主要通过实验室自动化、计算机应用和反应小型化实现。
- 技术:
- 焦磷酸测序(Pyrosequencing):通过测序过程中焦磷酸盐合成产生的发光来测量。
- Solexa 公司开发的荧光染料标记的边合成边测序技术,成为 Illumina 测序仪的基础。
- Roche 454 测序仪(2005):实现焦磷酸测序过程的完全自动化。
- SOLiD 系统(2007):连接测序。
- Ion Torrent(2011):通过检测新 DNA 合成时的氢离子进行测序。
- 优势:
- 化学试剂成本低。
- 可以处理稀疏材料。
- 高灵敏度检测低频变异,全面覆盖基因组。
- 样品多路复用,高容量。
- 可同时测序数千个基因。
- 局限性:
- 测序仪器昂贵,通常需要共享。
- 仪器体积大,不适合野外工作。
- 产生短测序片段(reads),难以用于新基因组组装。
- 测序结果质量依赖于参考基因组。
第三代测序 NGS
又称下一代测序技术,Next Generation Sequencing
- 背景:
- 第三代测序引入长读长测序和实时测序两项创新。
- 长读长测序对于没有参考基因组的新基因组组装非常重要。
- 实时测序结合便携式测序仪,使测序可以在野外进行。
- 技术:
- PacBio ZMW 测序(2010):
- 使用纳米孔(nanoholes)和单个 DNA 聚合酶。
- 通过荧光染料标记的核苷酸发出的荧光信号进行测序。
- 读长通常为 8-15 kb,最高可达 70 kb。
- Oxford Nanopore 测序(2012):
- 便携式测序仪(如 GridION、MinION、Flongle)。
- 通过检测核酸通过蛋白质纳米孔时的电流变化进行测序。
- PacBio ZMW 测序(2010):
- 优势:
- 长读长有助于组装大型新基因组。
- 便携式测序仪适合野外工作。
- 可直接检测 DNA 和 RNA 序列的表观遗传修饰。
- 测序速度快。
- 局限性:
- 一些第三代测序仪的错误率高于第二代测序仪。
- 试剂成本较高。
NGS 流程概述
- 样品和文库制备:
- 将 DNA 样品片段化,并连接接头分子,用于后续杂交和提供引发位点。
- 扩增和测序:
- 将文库转化为单链分子,并通过聚合酶链反应(PCR)等方法创建 DNA 分子簇。
- 每个簇在一次测序运行中进行独立反应。
- 数据输出和分析:
- 测序实验的输出取决于测序技术和化学成分。
- 数据量通常很大,需要复杂且计算密集的处理。
单细胞技术
单细胞多组学分析通过高通量技术对单个细胞的多种分子层(转录组、表观基因组、蛋白组等)进行联合分析,揭示细胞异质性、动态过程及调控机制。其核心在于整合RNA、ATAC、蛋白、空间信息等多模态数据,全面解析细胞状态和功能。
与传统的批量RNA测序技术(bulk RNA-seq)不同,单细胞RNA测序能够在单个细胞水平上揭示基因表达的异质性。其核心优势在于能够揭示细胞之间的差异,而这些差异在混合样本中往往被掩盖。
主要应用于:
- 细胞异质性解析(如肿瘤微环境)
- 发育分化机制(如干细胞命运决定)
- 调控网络构建(如转录因子互作)
单细胞技术的快速发展使得我们能够在单细胞水平上进行高通量的分子特征分析。除了传统的单细胞转录组测序(scRNA-seq),现在还可以测量染色质可及性、表面蛋白表达、适应性免疫受体库(TCR/BCR)以及细胞的空间位置等多模态数据。这些技术的发展推动了计算方法的创新,目前已有超过1400种工具可用于分析单细胞数据。
二、核心任务(Key Tasks)与实验流程
1. 单细胞转录组学(scRNA-seq)
背景
单细胞测序(scRNA-Seq)可揭示细胞类型特异性和细胞间相互作用。
为什么要做单细胞测序: “传统的Bulk RNA测序就像一杯混合果昔,只能看到整体的平均风味;而单细胞RNA测序则像品尝每一颗果实,能辨别不同细胞的独特特征与功能。”
实验流程
样本准备与文库制备
样品制备
- 组织采集:根据研究目标,选择合适的组织样本。例如,可以是动物组织、植物组织或临床样本。
- 细胞悬液制备:将组织样本处理成单细胞悬液。这一步骤需要根据样本类型选择合适的酶解或机械分离方法,以确保细胞的完整性和活性
单细胞分离
- 细胞分离技术:使用荧光激活细胞分选(FACS)、磁珠分选(MACS)或微流控技术等方法将单个细胞分离并分装到反应体系中。
- 细胞分选:将单细胞分配到微孔板、液滴或微流控芯片中,确保每个反应单元只包含一个细胞
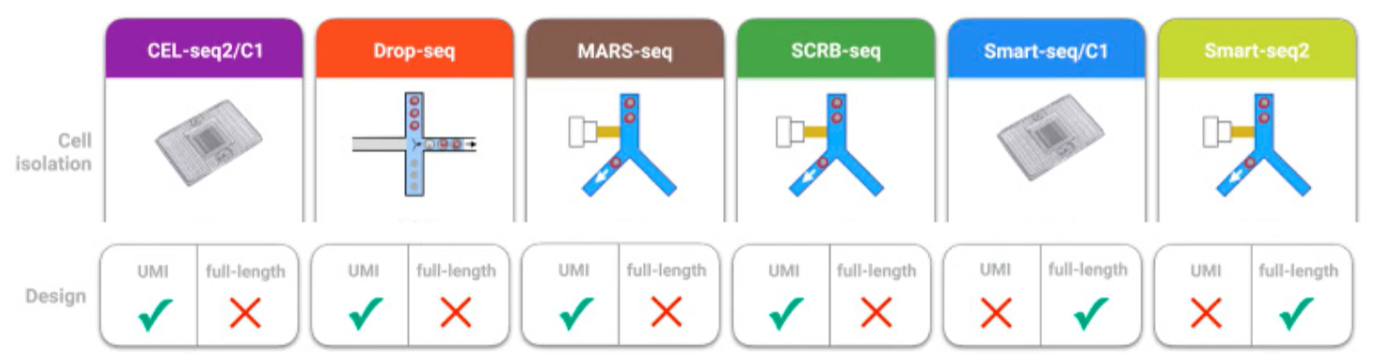
- 实验方法:目前有三种类型的单细胞测序协议(Single-cell sequencing protocols),主要根据细胞分离方式进行分组。
基于微流控装置
- 技术:
- 使用微流控装置将细胞封装在水凝胶液滴中,形成单细胞反应室,用于后续的裂解、逆转录和扩增等操作。
- 常见方案包括使用 Drop-seq、InDrop 和 10x Genomics Chromium。
- 优势:
- 成本低,可高通量测序大量细胞。
- 可使用 UMI。
- 局限性:
- 转录本回收率低(约 10%)。
- 仅捕获 3’ 或 5’ 末端,不覆盖全长转录本。
- 5’ 端:指的是核酸分子的5’磷酸基团所在的末端。在转录组测序中,5’端测序通常用于捕获转录本的起始部分,这对于研究基因的启动子区域、转录起始位点等具有重要意义。
- 3’ 端:指的是核酸分子的3’羟基所在的末端。3’端测序则侧重于捕获转录本的末端,常用于研究转录本的poly(A)尾结构、转录终止位点等
- 对比:
- 10x Genomics 在灵敏度和数据质量方面优于 Drop-seq 和 InDrop。技术噪声最低为10X Genomics,其次为Drop-seq、InDrop。
- Drop-seq 和 InDrop 更灵活,可调整和开源。
- 10X Genomics更喜欢捕获和放大较短的基因和GC含量较高的基因,而Drop-seq则更喜欢GC含量较低的基因。
- 尽管10X Genomics在各个方面都表现出色,但每个细胞的成本也大约是其他协议的两倍。
基于板的方案
- 技术:
- 将细胞物理分离到微孔板中,通过荧光激活细胞分选(FACS)或微量移液进行细胞分选。
- 每个孔作为一个独立的反应体系,进行后续的单细胞测序操作
- 常见方案包括 SMART-seq2、MARS-seq、QUARTZ-seq 和 SRCB-seq。
- 优势:
- 每个细胞可恢复更多基因,适合深度表征。
- 可在文库制备前收集细胞信息(如细胞大小、标签强度)。
- 可进行全长转录本恢复。
- 局限性:
- 实验规模受限于处理单元的通量。
- 碎片化步骤消除链特异性信息。
- 可能劳动密集型,存在技术噪音和批次效应。
Fluidigm C1
- 技术:
- 商用微流控芯片,自动将细胞加载到小型反应室中。
- 结合 RNA 提取和文库制备,自动化,减少人工操作。
- 优势:
- 全长转录本覆盖。放大步骤在单独的孔中进行,因此可以实现全长测序,有效减少了许多其他单细胞RNA测序协议的3’偏差。
- 可恢复剪接变体和 T/B 细胞受体库多样性。
- 局限性:
- 每个细胞成本高。
- 仅捕获约 10% 的细胞,不适合稀有细胞类型。
- 对细胞大小有偏倚。要求细胞混合度较均匀,因为细胞根据其大小在微流控芯片上会到达不同的位置,这可能引入潜在的位置偏差。
library preparation
- RNA提取:从单细胞中提取RNA,通常需要使用特殊的试剂和方法来避免RNA降解。
- cDNA合成:通过逆转录反应将RNA转录为cDNA,这一步骤通常需要加入唯一分子标识符(UMI)以标记原始RNA分子。
- 文库扩增:对cDNA进行PCR扩增,以获得足够的文库用于测序。
- 文库纯化:去除引物二聚体和其他杂质,确保文库质量
逆转录与扩增
- SMART技术合成cDNA
- UMI标记消除PCR偏差
- Unique Molecular Identifier 用于区分和追踪单个分子
- 基于液滴的 scRNA-seq 为多个细胞中的基因生成唯一分子标识符 (UMI) 计数,旨在确定每个基因和每个细胞的分子数量。它假设每个液滴都包含来自单个细胞的 mRNA
- 双细胞、空液滴和游离 RNA 可能违反这一假设。
- 消除PCR扩增偏差、提高计数精度、减少技术噪声
- 基于液滴的 scRNA-seq 为多个细胞中的基因生成唯一分子标识符 (UMI) 计数,旨在确定每个基因和每个细胞的分子数量。它假设每个液滴都包含来自单个细胞的 mRNA
- Unique Molecular Identifier 用于区分和追踪单个分子
进行测序
测序平台选择
根据实验需求选择合适的测序平台,如Illumina、10x Genomics、纳米孔测序等。
1.Illumina平台
- 技术原理: 基于边合成边测序(Sequencing by Synthesis, SBS) 技术。其核心原理是
- 将DNA片段固定在流动池(Flow Cell)的表面上。
- 通过添加荧光标记的核苷酸进行DNA合成,每次合成一个核苷酸时,会释放出特定的荧光信号。
- 通过光学检测系统记录荧光信号,从而确定DNA序列。
- 优势
- 高通量:能够同时测序数百万个DNA片段,适合大规模单细胞测序。
- 高准确性:测序错误率低,适合需要高精度的基因表达分析。
- 成熟的技术:Illumina平台技术成熟,数据质量稳定,有大量的分析工具和流程支持。
- 局限性
- 短读长:单次测序读长通常在100-300bp之间,对于复杂基因组或长转录本的分析可能不够全面。
- 成本较高:虽然单次运行成本相对较低,但设备和试剂费用较高,尤其是对于大规模实验来说成本较高。
- 测序运行:
- 文库制备:将单细胞RNA提取并逆转录为cDNA,构建测序文库。
- 流动池加载:将文库加载到Illumina流动池中,固定在流动池表面。
- 测序反应:在流动池中加入荧光标记的核苷酸,进行DNA合成反应。
- 数据采集:通过光学检测系统记录荧光信号,生成原始测序数据。
- 数据分析:将原始数据进行质量控制、比对和分析,得到基因表达谱等信息。
2.10x Genomics平台
技术原理:基于微流控技术和高通量测序,其核心原理是
- 使用微流控装置将单细胞封装在水凝胶液滴(GEMs)中,每个液滴包含一个细胞和一个带有条形码的凝胶珠。
- 细胞裂解后,RNA被逆转录为cDNA,并带有独特的条形码。
- 将所有液滴中的cDNA合并,构建测序文库。
- 使用Illumina平台进行测序,通过条形码区分不同细胞的转录本。
优势
- 高通量:能够同时处理大量单细胞,适合大规模单细胞测序。
- 成本低:单细胞测序成本较低,适合大规模实验。
- UMI支持:使用唯一分子标识符(UMI)进行分子计数,提高数据准确性。
- 成熟的解决方案:10x Genomics提供完整的实验和分析流程,易于操作和数据分析。
局限性
- 转录本覆盖不全:通常只捕获转录本的3’或5’末端,不覆盖全长转录本。
- 细胞捕获效率低:细胞捕获率较低,可能丢失稀有细胞类型。
- 依赖Illumina平台:需要结合Illumina平台进行测序,增加了实验复杂性。
测序运行
- 细胞封装:将单细胞与凝胶珠一起封装在水凝胶液滴中。
- cDNA合成:在液滴中进行细胞裂解和逆转录反应,生成带有条形码的cDNA。
- 文库构建:将所有液滴中的cDNA合并,构建测序文库。
- 测序:将文库加载到Illumina测序仪上进行测序。
- 数据分析:通过条形码区分不同细胞的转录本,进行数据比对和分析。
3.纳米孔测序平台
技术原理:基于单分子实时测序技术,其核心原理是
- DNA或RNA分子通过一个纳米孔(Nanopore),纳米孔两侧施加电压。
- 分子通过纳米孔时,会引起电流的变化,根据电流变化的模式确定碱基序列。
- 纳米孔测序可以直接对单链DNA或RNA进行测序,无需PCR扩增。
优势
- 长读长:单次测序读长可达数千甚至数万碱基,适合复杂基因组和长转录本的分析。
- 实时测序:测序过程实时进行,可以在测序过程中动态调整实验参数。
- 便携性:设备小巧,适合现场测序和小规模实验。
- 直接RNA测序:可以直接对RNA进行测序,无需逆转录步骤,保留RNA的修饰信息。
局限性
- 高错误率:单次测序的错误率较高,需要通过重复测序或计算方法进行校正。
- 成本高:纳米孔试剂成本较高,尤其是对于大规模实验来说成本较高。
- 数据复杂:需要结合其他测序平台(如Illumina)进行辅助分析,以提高数据准确性和可靠性。
测序运行
- 文库制备:将单细胞RNA提取并逆转录为cDNA,构建测序文库。
- 条形码分配:使用UMI或其他条形码技术标记每个细胞的转录本。
- 测序反应:将文库加载到纳米孔测序芯片上,DNA或RNA分子通过纳米孔进行测序。
- 数据采集:实时记录电流变化,生成原始测序数据。
- 数据分析:通过计算方法校正测序错误,结合其他平台数据进行综合分析。
数据生成与分析
数据生成
- 原始数据:FastQ文件(包含细胞/UMI条形码和转录本序列)。
- 比对与定量:STAR/Salmon比对参考基因组,生成细胞×基因UMI计数矩阵。
数据集选择
单细胞的数据来源可以是自己实验室的送测数据或者是网络开源数据。
在用到单细胞测序的文章中,Data avaliability中可以找到数据来源,大部分都会放在_Gene Experssion Omnibus(GEO)_上。
一些数据库:_PanglaoDB_、_Single Cell Atlas_
格式转换
上游数据处理:针对sra或者fastq文件,常用的软件有Sratoolkit,Cellranger等。
SRA Toolkit是由 美国国家生物技术信息中心 (NCBI)开发的一套工具和库,用于处理和访问 Sequence Read Archive (SRA)数据库中的数据。SRA Toolkit提供了多种工具,如prefetch、fasterq-dump等,用于从SRA数据库中下载、转换和验证序列数据。
Cell Ranger是由 10x Genomics 开发的一款单细胞RNA测序数据分析软件,主要用于处理从10x Genomics单细胞RNA测序平台(如 Chromium System )生成的数据。Cell Ranger的主要功能包括:序列比对、转录本拼接、质量控制、基因表达量分析、数据可视化。
10x 可以产生三个标准文件格式:matrix.mtx、genes.tsv和barcodes.tsv,分别存储了细胞的表达矩阵、基因名称和细胞条形码。这些文件可以使用R语言中的Read10X函数读取,并创建Seurat对象进行后续分析。
使用python:转为adata
1 | import scanpy as sc |
数据分析流程详见下。
数据分析
数据预处理
标准的预处理流程涉及特征选择步骤,旨在排除可能不代表样本之间有意义的生物变异的无信息基因。
质量控制
Quality Control
原因
单细胞数据存在技术噪声(如低质量细胞、双细胞、环境RNA污染),需过滤干扰因素确保分析可靠性。
目标
- 去除低质量细胞(膜破损或死亡细胞)
- 识别并去除双细胞(同一液滴含两个细胞)
- 消除环境RNA污染
步骤
去除低质量细胞
- 预处理的第一步。
背景:
当一个细胞检测到的基因数量少、计数深度低且线粒体计数分数高时,它的膜可能破损,这可能表明细胞正在死亡。由于这些细胞通常不是我们分析的主要靶标,并且可能会扭曲我们的下游分析,因此我们在质量控制期间会去除它们。
为了识别它们,我们定义了细胞质量控制 (QC) 阈值。细胞 QC 通常对以下三个 QC 协变量进行:
- The number of counts per barcode (count depth)
- The number of genes per barcode
- The fraction of counts from mitochondrial genes per barcode
低质量细胞通常表现为检测到的基因数量少、计数深度低和线粒体基因比例高。
步骤:
- 筛选feature中RNA基因表达量。(eg监测到基因表达量大于200的细胞)
- 筛选线粒体基因的占比,因为过高的线粒体基因表达可能是细胞受损和凋亡相关。通常的筛选阈值可以是5%或10% (但根据所做小提琴图,再进行测试,不损失大量细胞地选择设定)
- 筛选UMI>1000。UMI < 1000 可能意味着细胞捕获失败、RNA降解或检测灵敏度不足,容易被视为低质量细胞。
方法:
- 通过手动设置阈值或基于中位数绝对偏差(MAD)的自动过滤方法,可以识别并过滤这些细胞。
- 手动设置阈值:研究人员根据数据分布图(例如基因数量、总计数、线粒体基因比例)设定一个或多个阈值。细胞的这些特征如果超出设定的范围,就会被认为是低质量细胞并被移除。
- 基于中位数绝对偏差(MAD)的自动过滤方法:计算每个细胞的基因数量、总计数等指标的中位数和绝对偏差(偏离中位数的程度)。如果某个细胞的这些指标偏离中位数的距离超过设定的阈值,就被认为是低质量细胞并被去除。
- 优点是无需人为设置具体的阈值,能够自动适应数据中的异常值。
去除双细胞
背景:
- 双细胞/双峰doublets, 定义为在同一细胞条形码下测序的两个细胞(一个滴液中包含两个细胞)。
- 这违反了每个滴液只包含一个细胞的假设。
- 如果双峰由相同的细胞类型(但来自不同的个体)形成,则称为同型,否则称为异型。
- 同型双峰不一定能从计数矩阵中识别出来,并且通常被认为是无害的,因为它们可以通过细胞哈希或 SNP 来识别。因此,它们的鉴定并不是双峰检测方法的主要目标。
- 由不同细胞类型或状态形成的双峰称为异型。它们的识别至关重要,因为它们很可能被错误分类,并可能导致下游分析步骤失真。因此,双峰检测和删除通常是初始预处理步骤。
方法:
- 双峰可以通过其大量读取和检测到的特征来识别,也可以通过创建人工双峰并将其与数据集中存在的细胞进行比较的方法来识别。双峰检测方法的计算效率很高,并且有软件包可用于此任务。
- scDblFinder是一种基于人工合成双细胞的检测方法,提高下游分析的准确性。研究表明,scDblFinder在双细胞检测的准确性和计算效率方面优于其他方法。
- scDblFinder基于一种假设:双细胞的转录组是两个单独细胞转录组的结合。该方法通过比较实际测量的细胞与人工合成的双细胞(即将两个单细胞的RNA合并成一个虚拟的双细胞样本)之间的相似性来识别双细胞。
环境噪声修正
背景:
- 对于基于液滴的单细胞 RNA-seq 实验,稀释液中存在一定量的背景 mRNA,这些 mRNA 与细胞一起分布到液滴中并与它们一起测序。这样做的净效应是产生背景污染,该污染代表的表达不是来自液滴中包含的细胞,而是来自包含细胞的溶液。
- 环境RNA污染可能导致细胞类型特异性标记基因在其他细胞群体中被检测到,从而混淆细胞类型。
方法:
- 方法原理
- 在单细胞测序实验中,每个分子会被分配到一个特定的条形码(barcode),用于区分不同细胞来源的分子。
- 可以通过对条形码分配的分子数量进行排序,从分子数量非常少的条形码到分子数量非常多的条形码。
- 需要确定一个阈值(threshold),以区分真实细胞和非细胞(如空液滴或细胞碎片)。
- 直方图可以展示分子计数的分布情况,有助于确定合适的阈值。
- 散点图(2D scatter plots)可以展示基因数量与计数深度的关系,有助于识别细胞类型特异性的属性。
- SoupX和CellBender是两种常用的方法,分别基于“空滴”和贝叶斯模型来估计和去除环境RNA污染。
- SoupX是一种基于“空滴”的方法,它假设环境RNA在细胞的基因表达数据中表现为噪声,并且这个噪声在不同细胞群体之间是均匀分布的。SoupX通过从每个细胞的基因表达数据中去除这一“空滴”污染,来提高数据的质量。该方法通过计算细胞间的RNA污染程度,消除环境污染对分析结果的干扰。
- CellBender是另一种去除环境噪声的方法,它采用贝叶斯模型来估计并去除环境RNA的影响。CellBender通过对每个细胞的RNA数据进行建模,并推测出其中的环境噪声成分,再将其从数据中移除。通过这种方式,CellBender能够提高细胞类型的识别准确性,尤其在组织复杂的情况下表现良好。
示例代码
1 | # 计算质量控制指标 |
结果
- 过滤后的干净表达矩阵
- 细胞数减少,基因检测质量提升(如线粒体基因占比下降)
标准化
Normalization
原因
技术因素(如测序深度、基因长度)导致细胞间表达量不可比,需消除技术偏差。
- 测序深度:不同细胞的测序覆盖度可能不同。
- 测序覆盖度:测序获得的序列占整个靶序列如基因组序列的比例。
- 由于基因组中的高GC、重复序列等复杂结构的存在,测序最终拼接组装获得的序列往往无法覆盖所有的区域。
到目前为止,我们从数据集中删除了低质量细胞、环境 RNA 污染和双峰,数据以计数矩阵形式提供。这些计数代表 scRNA-seq 实验中分子的捕获、逆转录和测序。
这些步骤中的每一个,都为相同细胞的测量计数深度增加了一定程度的可变性,因此计数数据中细胞之间基因表达的差异可能仅仅是由于采样效应。这意味着数据集以及计数矩阵仍然包含变化很大的方差项。
UMI 数据的理论和经验建立模型是 Gamma-Poisson 分布,$Var[Y]=μ+αμ^2$
与均值 μ 和过离散的二次均值-方差关系 α 。因为 α=0 这是 Poisson 分布,它 α 描述了 Poisson 之上的额外方差。
目的
消除技术偏差(如测序深度、基因长度影响),使样本间可比。
方法
“归一化” 通常是先将每个细胞的计数值除以该细胞的总表达量(或总 UMI)并乘以一个 “标准化因子”(如 1 万),再取对数(LogNormalize)以降低表达量差异过大的影响。
| 方法 | 校正对象 | 适用数据类型 | 计算方式 | 适用场景 |
|---|---|---|---|---|
| TPM | 基因长度 + 测序深度 | 全长转录本(Full-length) | 归一化基因长度后计算每百万分比 | Smart-seq2、全长测序 |
| CPM | 测序深度(总计数) | 3’ 端富集数据 | 每个基因计数 / 细胞总计数 × 10⁶ | 10x Genomics、低异质性数据 |
| Scran | 细胞 RNA 总量(size factor) | 高异质性单细胞数据 | 预聚类 → 计算 size factor 归一化 | 组织数据、复杂生物系统 |
| 皮尔森残差 | 方差-均值依赖性 | 归一化后数据 | 计算标准化残差,提高统计建模效果 | PCA、聚类、降维 |
TPM(Transcripts Per Million)
- 目标:消除基因长度和测序深度对表达量的影响。
- 计算: $TPM_{gene} = \frac{\text{基因读段数} / \text{基因长度}}{\sum (\text{所有基因读段数} / \text{基因长度})} \times 10^6$
- 基因读段数(Read Counts)
- 测序仪检测到的比对到某个基因的DNA/RNA短片段数量
- eg若基因A有500个读段 → 表示该基因在细胞中被检测到500次
- 读段数越多 → 该基因在细胞中表达量越高
- 测序仪检测到的比对到某个基因的DNA/RNA短片段数量
- 基因长度(Gene Length)
- 基因在DNA上的物理长度(单位:碱基数,bp)
- eg人类β-肌动蛋白基因长度约3,500 bp
- 关键影响:在全长转录本测序中,长基因更容易被随机打断 → 产生更多读段数
- 基因在DNA上的物理长度(单位:碱基数,bp)
- 总读段数
- 指单个细胞中所有基因的读段数之和(即该细胞的文库大小)
- eg某细胞检测到GeneA=100 reads,GeneB=200 reads,其他基因总和=700 reads → 总读段数=1000
- 指单个细胞中所有基因的读段数之和(即该细胞的文库大小)
- 基因读段数(Read Counts)
- 适用场景:
- 全长转录本测序(Full-length RNA-seq),如 Smart-seq2。
- 基因长度影响较大,需要进行校正。
CPM(Counts Per Million)
- 目标:校正测序深度,使细胞间的基因表达量可比。
- 计算: $CPM_{gene} = \frac{\text{基因计数}}{\text{细胞总计数}} \times 10^6$
- 适用场景:
- 适用于 3’ 端富集测序(如 10x Genomics)。
- 假设不同细胞的 RNA 总量相似,适用于克隆细胞系等低异质性数据。
Scran
- 目标:处理单细胞 RNA-seq 数据中细胞 RNA 总量差异的问题(如高RNA含量的巨噬细胞 vs 静息T细胞),使不同细胞的表达水平可比。
- 数据特征:存在细胞亚群间RNA总量的系统性差异
- 原理:
- 细胞可能有不同的 RNA 总量(如 T 细胞 vs 巨噬细胞)。
- 通过 预聚类 分组 → 计算 size factor → 归一化。
- 计算: $\text{SizeFactor}_i$ = ${median}g \times \frac{x{g_i}}{s_g}$
- $x_{g_i}$:基因 g 在细胞 i 的原始计数
- $s_g$:基因 g 在所有细胞中的计数中位数
- 适用场景:
- 适用于 细胞 RNA 总量差异较大的数据(如复杂组织样本)。
- 适用于 细胞异质性较高 的数据集。
皮尔森残差(Pearson residuals)
Pearson残差是一种基于标准化的统计量,用来评估细胞表达数据与期望值之间的差异。这种方法可以帮助消除细胞之间的技术性差异,使得不同细胞的表达谱具有可比性。
$Pearson残差 = \frac{观测值 - 期望值}{\sqrt{期望值}}$
这种归一化技术的动机是观察到 scRNA-seq 数据中的细胞间差异可能被具有技术效应的生物异质性混淆。它将计数深度显式添加为广义线性模型中的协变量。这种方法消除了技术的影响,同时保留了数据集中的细胞异质性。
- 目标:去除方差-均值依赖性,使数据更适合 PCA、聚类等分析。
- 计算:
- 计算每个基因-细胞组合的标准化残差: $R_{gi} = \frac{x_{gi} - \mathbb{E}[x_{gi}]}{\sqrt{\mathbb{V}[x_{gi}]}}$
- $x_{gi}$:基因 g 在细胞 i 中的原始计数
- $\mathbb{E}[x_{gi}]$]:期望值(基因的均值)
- $\mathbb{V}[x_{gi}]$:方差
- 计算每个基因-细胞组合的标准化残差: $R_{gi} = \frac{x_{gi} - \mathbb{E}[x_{gi}]}{\sqrt{\mathbb{V}[x_{gi}]}}$
- 适用场景:
- 主要用于 PCA、聚类、降维。
- 使变换后的数据更加正态化,减少高表达基因的主导效应。
示例代码
1 | # 使用CPM标准化 |
结果
- 标准化后的表达矩阵(如log(CPM+1))
- 高表达基因主导性降低,细胞间表达可比性增强
批次校正
Batch Correction
原因
- 批次效应是指在不同实验批次中由于操作、设备或环境等因素引入的非生物学差异。
- 例如,不同实验室采用不同的细胞解离方法,可能导致细胞表达不同的应激相关基因(如 JUN、JUNB、FOS 等),即使这些细胞在原始组织中具有相同的基因表达特征。
- 不同实验批次之间的差异也需要进行批次效应校正以去除非生物学差异。
目标
消除因实验批次、技术差异导致的非生物学变异。
批次效应识别
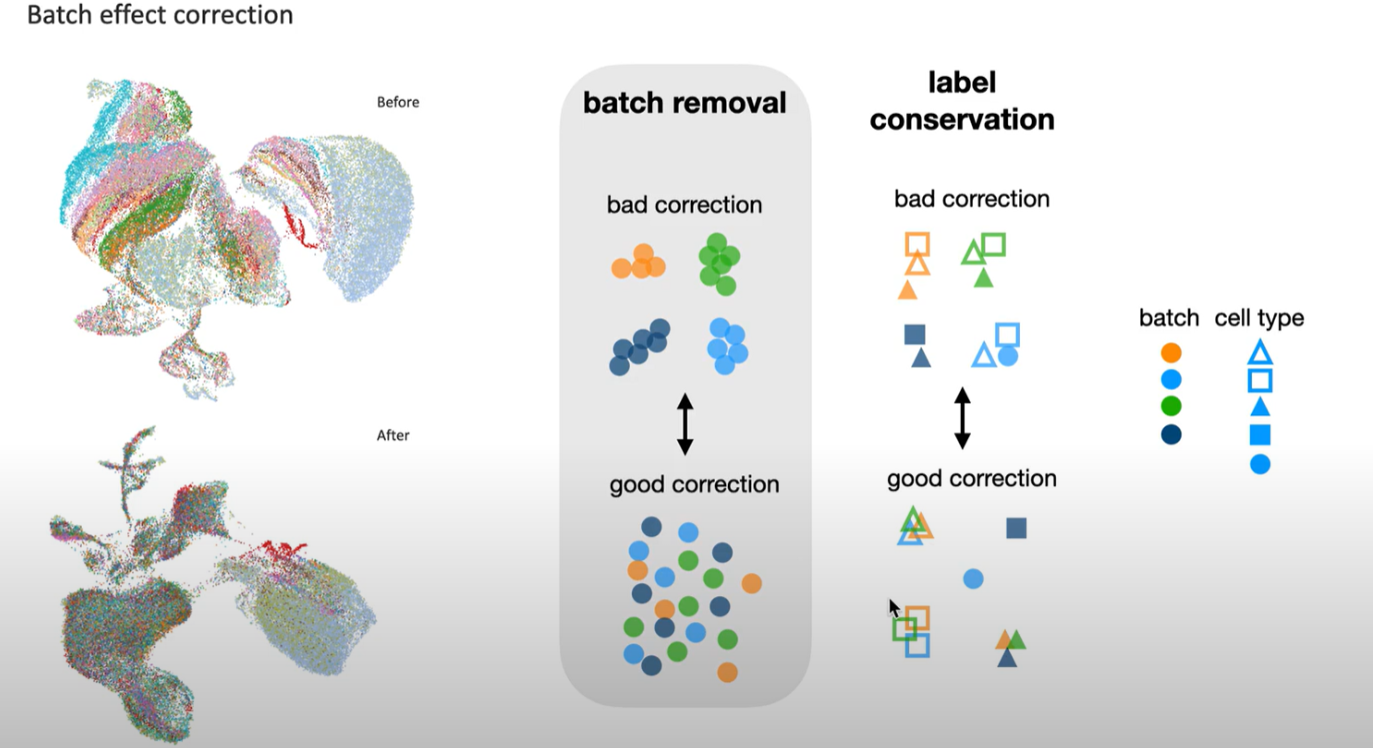
- 典型表现
- 批次在降维图(如PCA/UMAP)中,不同实验批次的细胞形成分离的簇状结构
- 如上图中”鸡胸”结构,生物学信号(如细胞类型)被批次主导
- 批次在降维图(如PCA/UMAP)中,不同实验批次的细胞形成分离的簇状结构
- 本质原因
- 技术差异(如不同试剂、操作人员)导致非生物学变异,属于数据系统性偏差
任务
- 去除 scRNA-seq 数据中的批量效应可分为两个子任务:
- 批量校正(batch correction)
- 批量校正处理同一实验中样本之间的批量效应,通常具有准线性效应;
- 数据集成(data integration)
- 而数据集成处理不同数据集之间的复杂批量效应,可能涉及不同的实验协议和细胞身份的差异。
- 批量校正(batch correction)
方法
主要使用Harmony和BBKNN。
常用方法可分为四类:
全局模型(Global models)
- 批量校正方法
- 将批量效应建模为所有细胞的一致(加法和/或乘法)效应,例如 ComBat。
eg - ComBat
- 输出形式:校正后的基因表达矩阵。
- 特点:基于贝叶斯框架,通过调整数据的批次效应,使得不同批次的数据在统计分布上更加一致。适用于已经被过滤和标准化后的数据。
- 参数方法(par.prior=TRUE):假设数据的批次效应可以通过正态分布的参数(均值和方差)来调整。这种方法在调整批次效应时没有考虑其他协变量,是一个纯粹的批次效应调整。
- 非参数方法(par.prior=FALSE):不假设批次效应的参数分布,而是直接估计和调整每个批次的效应,更加灵活地适应各种数据分布。这种方法只考虑批次间的均值差异,不调整方差,适用于批次间方差相似,但均值有偏差的情况。
- 适用场景:适用于需要保留基因维度进行下游差异分析的场景,尤其是批次效应主要体现在全局分布上的数据。
- 分类:全局模型,因为它通过全局调整数据的分布来去除批次效应。
线性嵌入模型(Linear embedding models)
批量校正方法
使用奇异值分解(SVD)的变体嵌入数据,通过低维嵌入空间进行校正。通过查找跨批次的相似细胞局部邻域来校正批量效应,如 Seurat、Scanorama、Harmony 等。
egSeurat
- 输出形式:输出校正后的表达矩阵。
- 特点:基于互最近邻(MNN)的思想,通过锚点对齐不同数据集。
- MNN(Mutual Nearest Neighbors):通过匹配相互最近邻来校正批次效应。不依赖于各个批次之间预定义或相等的群体组成。
- 适用场景:适用于需要保留基因维度进行下游差异分析的场景,尤其是批次效应主要体现在局部结构上的数据。
- 使用方法:
- 数据准备:将 AnnData 转换为 Seurat 对象。
- 找到锚点:使用
FindIntegrationAnchors()函数。 - 数据集成:使用
IntegrateData()函数。 - 可视化:通过 UMAP 嵌入展示校正效果。
Scanorama
- 输出形式:校正后的基因表达矩阵。
- 特点:基于谱聚类的方法,通过构建一个批次无关的低维空间来整合数据。保留基因维度,适合下游差异分析。
- 适用场景:适用于需要保留基因维度进行下游差异分析的场景,尤其是大规模数据集。
Harmony
- 输出形式:校正后的基因表达矩阵。
- 特点:基于迭代优化算法,通过全局信息去除批次效应。速度快、效果好,尤其在处理高维数据时表现良好。
- 适用场景:适用于大规模数据集,需要保留基因维度进行下游差异分析的场景。
基于图的方法(Graph-based methods)
批量校正方法
使用最近邻图表示数据,通过图结构进行校正。通过强制不同批次细胞之间的连接并修剪强制边来校正批量效应,例如 BBKNN、Conos。
egBBKNN(Batch Balanced K-Nearest Neighbors)
- 输出形式:细胞邻接关系图(非矩阵)。
- 特点:通过构建一个批次平衡的K近邻图来整合数据。高效处理大数据,保留局部结构。
- 适用场景:适用于百万级细胞数据集,需要高效处理大数据并保留局部结构的场景。
- 使用方法:
- 参数选择:根据细胞数量选择每批次的邻居数。
- 构建图:使用
bbknn.bbknn()替代常规的 KNN 图构建。 - 可视化:通过 UMAP 嵌入展示校正效果。
Conos
- 输出形式:细胞邻接关系图(非矩阵)。
- 特点:通过构建一个批次无关的图来整合数据。适用于大规模数据集,保留局部结构。
- 适用场景:适用于百万级细胞数据集,需要高效处理大数据并保留局部结构的场景。
深度学习方法(Deep learning approaches)
批量校正方法、数据集成方法
基于自动编码器网络,如 scVI、scANVI,通过条件变分自动编码器(CVAE)或嵌入空间中的局部线性校正来去除批量效应。
eg:scVI(单细胞变分推理)
- 输出形式:校正后的基因表达矩阵或低维嵌入。
- 特点:基于条件变分自动编码器(CVAE),直接对原始计数进行建模,适用于大规模数据集,平衡批量校正和生物变异保留。scVI的全面性、可扩展性好于scAVI。
- 适用场景:适用于需要保留基因维度进行下游差异分析的场景,尤其是大规模数据集。
- 使用方法:
- 数据准备:使用
scvi.model.SCVI.setup_anndata配置数据。 - 模型训练:通过
model_scvi.train()训练模型。 - 提取嵌入:使用
model_scvi.get_latent_representation()获取校正后的低维嵌入。 - 可视化:通过 UMAP 嵌入展示校正效果。
- 数据准备:使用
scANVI(单细胞注释变分推理)
- 输出形式:校正后的基因表达矩阵或低维嵌入。
- 特点:扩展了 scVI,结合细胞身份标签信息,更好地保留细胞类型差异。主要专注于半监督的细胞类型注释,即利用部分已知的细胞类型标签来推断其他细胞的状态
- 适用场景:适用于有细胞标签且需要保留生物变异的场景。
- 使用方法:
- 数据准备:与 scVI 类似,但需要提供细胞标签信息。
- 模型训练:通过
model_scanvi.train()训练模型。 - 提取嵌入:与 scVI 相同,但可能在保留生物信号方面表现更好
特征选择
Feature Selection & Highly variable Genes Selction
原因
单细胞数据高维度(~20,000基因)但多数为噪声,需筛选信息基因。
现在有一个标准化的数据表示,它仍然保留了生物异质性,但降低了基因表达的技术采样效应。
单细胞 RNA-seq 数据集通常包含多达 30,000 个基因,到目前为止,我们只删除了至少 20 个细胞中未检测到的基因。然而,在scRNA-seq实验中,通常只有一小部分基因是信息丰富且生物学上可变的。许多剩余的基因没有信息,并且大多包含零计数。
传统方法通过计算所有基因的变异系数或平均表达水平来选择500-2000个基因用于下游分析。然而,这些方法对之前的归一化技术非常敏感。因此采用以下特征选择方法:
特征选择是选择最具有信息量、变化最大或最偏差的特征子集的过程。
目标
识别高变基因(HVGs)用于下游分析。
筛选Highly variable Genes方法
高变基因(Highly Variable Genes, HVGs)
在上万基因中,并不是所有基因都对细胞分群或异质性有贡献;许多基因只是在低水平稳定表达,没有提供多少区分度。
“高变基因(HVG)” 指在不同细胞之间表达变异度(变异、平均值等统计)显著高于其他基因的基因。它们往往富含能区分细胞类型或状态的重要信息。
其表达水平变化较大,表达量在不同细胞之间存在显著差异,通常用于描述细胞类型之间的异质性。
高变基因的特征包括:
- 集中在低表达基因:大多数高变基因的表达水平较低。
- 方差大于预期:高变基因的方差通常大于负二项模型预估的方差。
- 较高的零值比例:高变基因在某些细胞中可能完全不表达
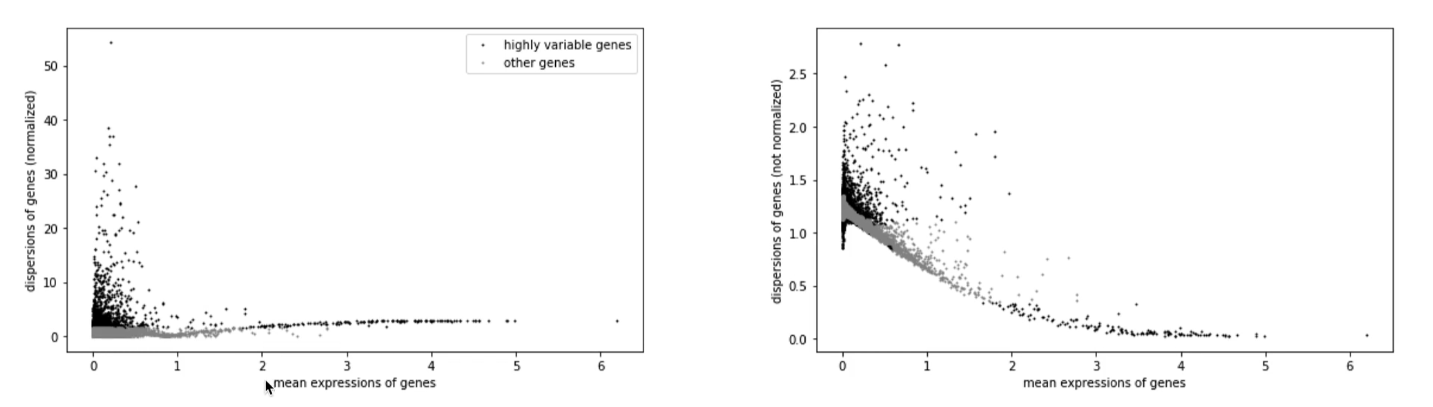
目的:聚焦携带生物学变异的基因,降低噪声。从2万+基因中筛选出500-2000个携带真实生物学变异的基因。
筛选策略
- 核心原理:
- 离散度(Dispersion) = 基因表达的方差 / 均值
- 筛选标准:离散度高且平均表达适中的基因(排除技术噪声)
- 核心原理:
筛选工具:Seurat
原理
- ① 按平均表达分箱 → ② 计算各箱内离散度 → ③ 按标准化离散度筛选
定位
- 单细胞分析最流行的R语言工具包
偏差特征选择
Germain等人提出了一种使用偏差进行特征选择的方法,该方法适用于原始计数。
偏差可以封闭形式计算,用于量化基因是否在细胞中显示出恒定的表达谱。具有高偏差值的基因表明它们不符合零模型(即,它们在细胞中的表达不是恒定的)。根据偏差值,该方法对所有基因进行排名,并选择高度偏差的基因。
示例代码
1 | sc.pp.highly_variable_genes(adata, min_mean=0.0125, max_mean=3, min_disp=0.5) |
结果
- 保留500-2000个HVGs(如免疫数据中的CD3E、CD79A)
降维
原因
高维数据难以可视化/聚类,需压缩至低维保留主要结构。
目标
- 降维:从成千上万的基因维度浓缩到几十或更少的主成分(PC);降维至2-50维(如PCA的Top 50 PCs)用于后续分析。
- 捕捉主要的差异:首几个 PC 往往能解释主要的表达变异,后面的 PC 可能主要是噪音;
- 加速后续分析:如 t-SNE/UMAP/ 聚类时,直接在 PC 空间构建相似度图,速度更快,且更能集中在关键信号上。
方法
- 进行数据缩放,让各个基因表达呈现在统一尺度。
- 对每个基因进行线性变换,让它在所有细胞上的平均表达量变为 0,标准差变为 1。
- 选定PC数
- 选定 PC 数:以 ElbowPlot 或其他方法
- 目的:避免盲目使用太多 PC 导致噪音进入;也避免只用过少 PC 可能丢失重要信息;在单细胞中常选取前 10 - 30 个 PC;原文若指定前 10 个 PC,则是作者根据自己数据分布做过考量并发现 10 个 PC 足够。
- 原理:PCA 会给出很多 PC(与基因数或细胞数相关),但通常只有前若干个 PC 是真正反映主要生物学差异的。
- ElbowPlot(肘部图)通过显示每个 PC 的标准差或方差贡献度,你可以直观地看到 “拐点”(像手肘形状),拐点前的 PC 贡献大,拐点后的 PC 贡献小而可能是噪音。
- 使用PCA、t-SNE、UMAP等方法进行降维,便于数据可视化和后续分析。
ElbowPlot eg: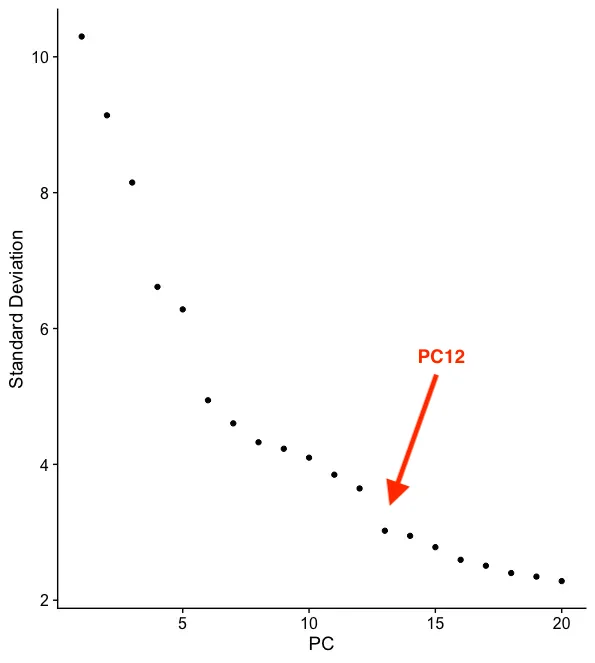
PCA (Principal Component Analysis)
主成分分析(PCA)是一种经典的降维方法,试图寻找数据中方差最大的方向(即主成分,PC1、PC2、PC3…),并将高维的基因表达矩阵投影到这些主成分上。它通过线性变换将数据投影到一个新的坐标系,保留数据的最大方差。
PCA 适用于大规模数据的初步降维,能够减少数据的噪音并保留最重要的信息。
具体做法通常是对 “(细胞 × 高变基因)” 矩阵做奇异值分解(SVD),先得到一个协方差矩阵,再提取若干最大的特征值 / 特征向量(主成分)。
t-SNE (t-Distributed Stochastic Neighbor Embedding)
t-SNE 是一种非线性降维方法,主要用于数据可视化。t-SNE 通过将相似的点映射到低维空间中的相近位置,适合于展示群体结构。它的缺点是计算速度较慢,且对于大规模数据可能不太适用。
UMAP (Uniform Manifold Approximation and Projection)
UMAP(Uniform Manifold Approximation and Projection)是一种基于流形学习的降维方法,能够保留数据的局部和全局结构。
它相对于 t-SNE 更加高效且适用于大数据集,常用于高维数据的可视化和聚类分析。
通过UMAP嵌入,可以将高维的基因表达数据映射到二维或三维空间中,便于观察细胞之间的相似性和差异性。
- 它通过以下步骤实现降维:
- 局部邻域保留:UMAP计算每个点的局部邻域,并将这些邻域嵌套在超平面中,从而保持原数据中的局部结构。
- 相似性度量:UMAP使用非线性变换来保持邻近点之间的相似度,能够捕捉数据中的复杂结构。
- 维度嵌入:UMAP在低维空间中对点进行重排,使得低维空间的距离大致对应原数据中的相似性
结果
低维嵌入(如UMAP图),细胞按类型/状态聚集
识别细胞结果
预处理和可视化使我们能够描述我们的 scRNA-seq 数据集并降低其维度。到目前为止,我们嵌入和可视化了单元格,以了解数据集的基本属性。然而,它们的定义仍然相当抽象。
单细胞分析的下一个自然步骤是识别数据集中的细胞结构。
Clustering
聚类分析
聚类是无监督机器学习中常见的问题,通过最小化简化表达空间中的簇内距离来推导簇。在scRNA-seq数据分析中,我们通过聚类来推断相似细胞的身份。
KNN图
KNN图由反映数据集中细胞的节点组成。我们首先计算所有细胞在PC简化表达空间上的欧几里得距离矩阵,然后将每个细胞连接到其K个最相似的细胞。K通常设置在5到100之间,具体取决于数据集的大小。
Leiden算法
Leiden算法是Louvain算法的改进版本,适用于单细胞RNA-seq数据分析。它通过考虑集群中细胞之间的链接数与数据集中总体预期链接数来创建集群。
Leiden模块有一个分辨率参数,允许确定分区集群的规模,从而确定聚类的粗糙度。分辨率参数越高,聚类越多。
- 控制簇的粒度:低分辨率→大簇;高分辨率→细粒度小簇(示例:PBMC数据中的不同分辨率结果)。
结果)。
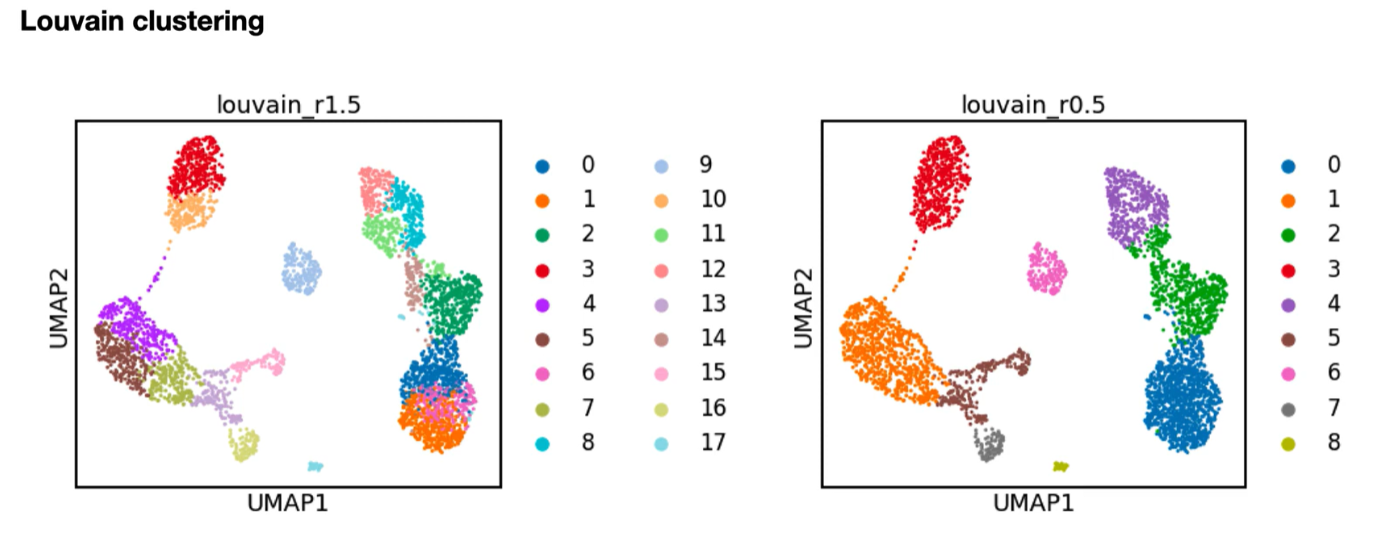
- 调参经验:根据生物学问题调整(如区分主类 vs. 亚型)。
Annotation
- 注释:结合自动化注释工具(如CellTypist、Clustifyr)和手动注释方法,利用差异表达基因(marker genes)进行细胞类型注释。
原因
低维聚类结果需关联生物学意义(如细胞类型、状态)。
为了更好地了解数据并利用现有知识,重要的是要弄清楚数据中每个细胞的“细胞身份”。
根据已知(或有时未知)细胞表型标记数据中细胞组的过程称为“细胞注释”。
目标
为每个簇分配细胞类型标签。
方法
有很多方法可以注释细胞(例如,基于批次、疾病、性别、细胞类型等)
手动注释策略
- 参考已知标记基因(如免疫细胞:CD3E→T细胞,CD19→B细胞)。
- 结合文献和数据库(如CellMarker, PanglaoDB)。
- 检查标记的“特异性”与“敏感性”:避免广泛表达基因(如线粒体基因)。
自动注释工具
训练分类器进行自动注释
- 自动工具(CellTypist)
- 预训练分类器匹配公共参考数据集
- Garnett:基于预定义标记基因集的分类器。
- 优点:快速批处理;缺点:依赖先验知识,可能遗漏新类型。
注释图数据解读
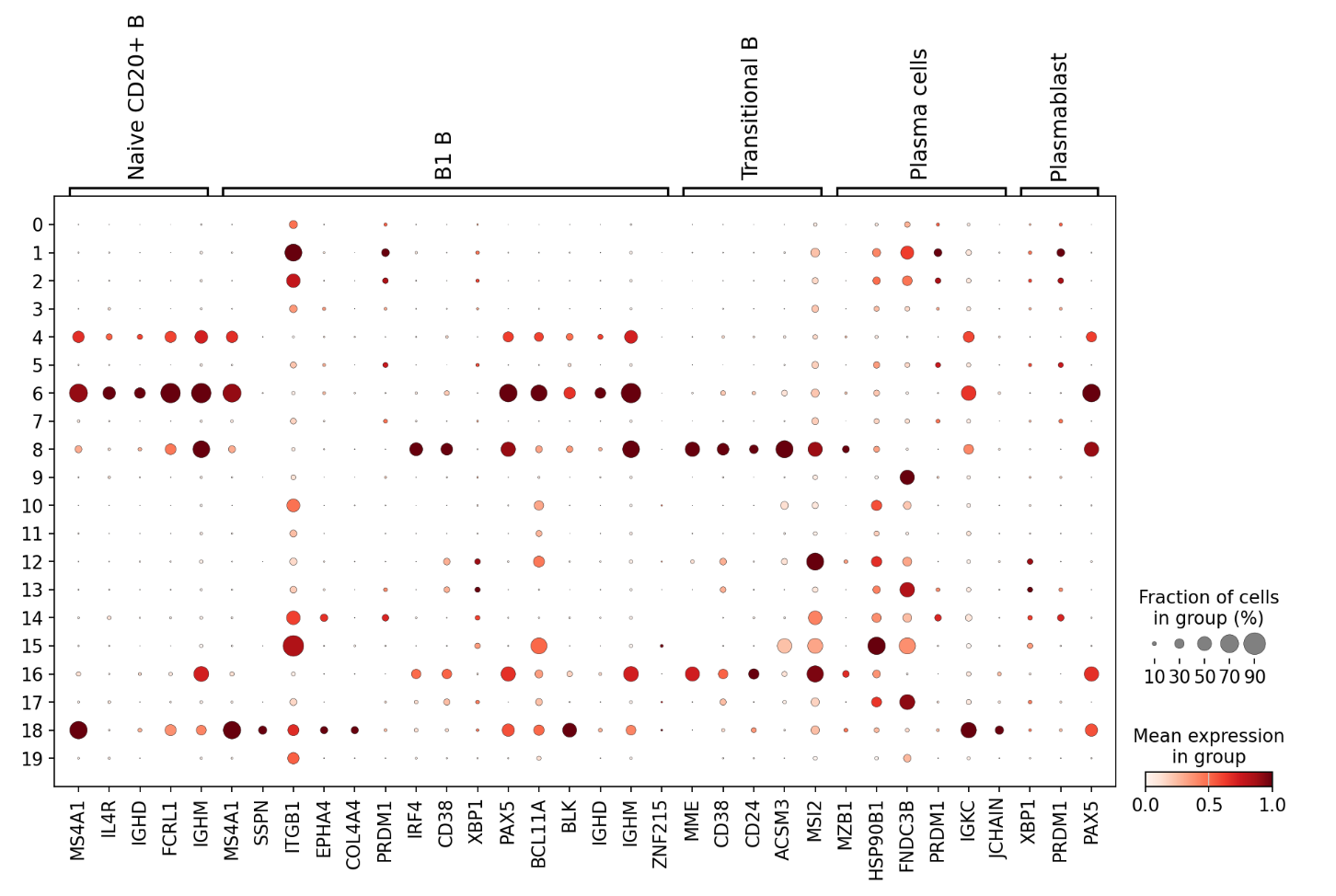
- 横轴(X轴):列出了多个基因的名称,包括MS4A1、IL4R、IGHD等,这些都是与免疫反应和B细胞功能相关的基因。
- 纵轴(Y轴):表示B细胞的不同发育阶段,从左到右依次为:
- Naive CD20+ B(初始B细胞)
- B1 B(早期B细胞)
- Transitional B(过渡阶段B细胞)
- Plasma cells(浆细胞)
- Plasmablast(浆母细胞)
- 点的颜色:代表基因在该细胞群体中的平均表达水平。颜色从浅到深,表示表达水平从低到高。图例中的颜色条显示了表达水平的范围,从0(最浅)到1(最深)。
- 点的大小:表示在该细胞群体中表达该基因的细胞比例。图例中显示了不同大小的点对应的细胞比例,从10%(最小)到90%(最大)。
- 数据解读:
- 在Naive CD20+ B阶段,可以看到一些基因如IGHM、IGHD等有较高的表达水平(颜色较深)。
- 在B1 B阶段,基因如FCRL1、MS4A1的表达水平较高。
- 在Plasma cells和Plasmablast阶段,可以看到一些与浆细胞功能相关的基因如XBP1、PAX5等有较高的表达水平。
- 通过比较不同阶段的点的颜色和大小,可以观察到基因表达的变化趋势,例如某些基因在特定发育阶段表达增加或减少。
下游任务
轨迹推断
Inferring trajectories
- 轨迹推断:使用PAGA、RaceID/StemID等工具推断细胞分化轨迹。
原因
用以研究细胞状态转换(如分化、激活)。
传统的单细胞RNA测序(scRNA-seq)无法追踪细胞的发育过程,因为测序过程会破坏细胞。尽管有新技术可以按顺序记录转录组,但这些技术在实验上具有挑战性且难以扩展到更大的数据集。
目标
构建伪时间轴,排序细胞发育阶段。
伪时间排序
Pseudotemporal Ordering
伪时间排序是指将细胞按照其在发育或分化过程中的相对顺序进行排列,从而重建细胞状态的动态变化过程。
- 核心目标:
- 为每个细胞分配一个“伪时间”值,表示细胞在发育轨迹中的相对位置。即通过将细胞状态映射到一个连续的域(伪时间)来重建细胞发育的轨迹。
- 工作流程:
- 伪时间根据细胞在发育过程中的阶段对细胞进行排序,不成熟的细胞分配较小的值,成熟的细胞分配较大的值。
- 应用场景:
- 适用于需要明确细胞发育顺序的研究,例如细胞分化过程、疾病进展等。
- 伪时间排序的结果通常用于可视化细胞状态的变化,分析基因表达的动态变化
伪时间构造
Pseudotime Construction
伪时间构造是指通过一系列计算方法构建出细胞的伪时间轨迹,包括从高维数据中提取低维表示、构建细胞之间的连接关系、定义细胞状态的转换路径等。
- 工作流程:伪时间构造通常包括将高维数据投影到低维表示,并基于低维数据构建伪时间。
- 常用方法:
- 聚类方法(Cluster Approach):首先对细胞进行聚类,然后根据相似性或最小生成树(Minimum Spanning Tree, MST)连接聚类,从而构建伪时间。
- 图方法(Graph Approach):通过构建基于低维表示的图来定义聚类和排序,例如PAGA(Partition-based Graph Abstraction)。
- 基于分区的图抽象(Partition-based Graph Abstraction)
- 核心思想:
- 分区(Partition):先将单细胞数据聚类为多个细胞群(簇),每个簇代表一个细胞亚群(如T细胞、B细胞)。
- 图抽象(Graph Abstraction):在簇级别构建图结构,节点为簇,边表示簇间的连接强度或过渡可能性。
- 典型方法:PAGA(Partition-based Graph Abstraction)
- 步骤:
- 使用聚类算法(如Leiden)将细胞分群。
- 计算簇间连接强度:基于原始单细胞图中跨簇的边数量或相似性得分。
- 构建抽象图:簇为节点,边权重反映簇间关系(如连接数占比)。
- 优势:
- 揭示全局拓扑结构(如分支、循环),避免高维噪声干扰。
- 支持复杂轨迹分析(如多分支分化、细胞周期)。
- 输出示例:
- 节点大小:代表簇内细胞数量。
- 边粗细:反映簇间连接强度。
- 边权重计算:
- 在PAGA中,边权重通常基于原始单细胞图中跨簇边的比例,例如:$\text{Weight}_{A \rightarrow B} = \frac{\text{跨簇边数(A到B)}}{\text{簇A内边数} + \text{簇B内边数}}$
- 步骤:
- 基于分区的图抽象(Partition-based Graph Abstraction)
- 流形学习方法(Manifold-learning Based Approaches):使用主曲线或图来估计底层轨迹,例如Slingshot。
- 主曲线是一种一维曲线,能够连接高维空间中的细胞观测值。Slingshot通过拟合主曲线来捕捉细胞状态之间的连续变化。
- Slingshot将细胞聚类后,通过主曲线连接这些聚类,从而构建出细胞发育的轨迹。
- 沿着估计的轨迹为每个细胞分配伪时间值,反映细胞在发育过程中的相对位置。
- 概率框架(Probabilistic Frameworks):为有序的细胞对分配转换概率,定义随机过程以构建伪时间,例如扩散伪时间(Diffusion Pseudotime, DPT)和Palantir。
- DPT:基于扩散过程(diffusion process),通过计算细胞之间的扩散距离来推断细胞的发育顺序。
- Palantir:基于马尔可夫链,计算细胞之间的转移概率,识别细胞的终末状态。
结果
- 轨迹图(如干细胞→祖细胞→终末分化细胞)
eg: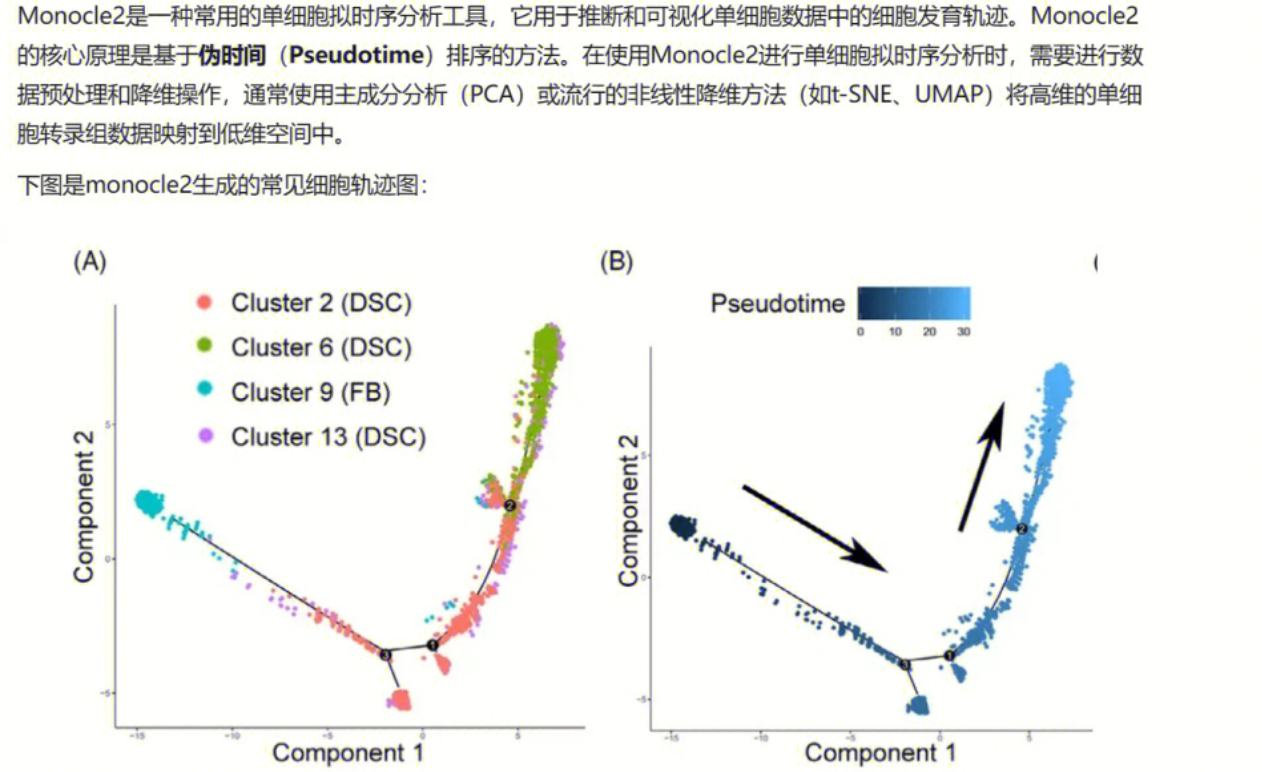
图中每个圆点代表一个细胞,其不同的分支代表具有不同细胞命运和转录组表达模式的细胞亚群。左图不同颜色代表了不同细胞 Cluster,右图其中颜色由深到浅为伪时间的顺序(这个伪时间值表示了细胞在发育过程中的相对顺序,而不是实际时间)。
展望
- 方法选择:选择合适的轨迹推理方法需要了解生物过程的性质(线性、循环或分支)。
- 新技术的应用:随着测序技术的进步,新的信息源(如ATAC-seq、CITE-seq、DOGMA-seq、RNA速度等)为更准确和稳健地估计轨迹和伪时间提供了可能。
RNA 速度(RNA Velocity)分析
原因
用以预测细胞未来状态,揭示动态过程。
在传统的单细胞转录组学分析中,通常只能获取单一快照,即某一时刻的基因表达数据。这意味着我们只能看到基因在该时刻的表达状态,但无法追踪细胞在时间上的变化或了解基因表达未来的走向。
单细胞数据集能够以高分辨率研究生物过程。然而,由于单细胞测序方案的破坏性,无法对同一细胞进行多次测序以追踪其表型特征随时间的变化。此外,实验技术不仅无法测量不同时间点的细胞特征,也无法测量这些变化发生的速度。
虽然轨迹推理(Trajectory Inference, TI)工具可以恢复细胞在发育过程中的时间位置,但经典 TI 方法缺乏动态信息。
RNA Velocity(RNA速度)作为一种新的分析方法,旨在通过观察基因表达的转录动态,打破单一快照的限制。RNA速度能够从转录组数据中推断出细胞的未来状态,进而揭示细胞在转录过程中的”速度”和”趋势”。
原理
RNA速度分析的核心是通过建模来推断细胞中基因表达的动态变化方向和速度。
基本逻辑:利用未剪接的pre-mRNA和剪接后的成熟mRNA的相对比例,来推断基因表达的变化趋势。
单细胞测量是快照数据,无法与时间作图。经典的 RNA 速度方法依赖于研究每个基因的未剪接和剪接 RNA 的细胞特异性元组,形成相位画像(phase portrait)。
具体来说,未剪接mRNA的相对增加表示基因正在上调(诱导),而未剪接mRNA的相对减少则表示基因正在下调(抑制)。RNA速度建模的目的是通过数学模型来描述这一动态过程,并推断出细胞在发育或分化过程中的“速度”和方向。
细胞转录组谱的变化由一系列事件触发:DNA 被转录生成未剪接前体信使 RNA(pre-mRNA),其中包含外显子(exons)和内含子(introns)。内含子被剪接去除后形成成熟的剪接 mRNA。单细胞 RNA 测序(scRNA-seq)可以区分未剪接和剪接的 mRNA 读数。通过识别未剪接和剪接的读数,可以建立描述剪接动力学的动态模型,并根据单细胞数据推断模型权重。模型描述的剪接 RNA 的变化称为 RNA 速度。
- 基因结构:
- 外显子(Exons) 和内含子(Introns) 构成了基因的基本结构。外显子是基因编码的部分,而内含子则是被剪接掉的部分。
- RNA的成熟与剪接:
- 成熟RNA:从外显子中转录出来的RNA,经过剪接后形成成熟的mRNA,可以用于蛋白质合成。
- 未成熟RNA:含有内含子的RNA(未剪接的部分),这种RNA需要经过剪接处理才能变为成熟的mRNA。
- RNA速度如何反映转录动态:
- RNA速度基于“未剪接”与“剪接”mRNA的比例差异进行推断。
- 当一个基因被激活时,未剪接的mRNA比例较高,而当转录停止时,未剪接mRNA的比例下降,剪接mRNA比例上升。
- 实验方法与技术:
- 通过不同的实验协议(如SMART-Seq与10x Genomics)可以观察到不同的未剪接和剪接mRNA比例,从而反映基因的转录动态。
参数推断(Parameter Inference)
假设转录、剪接和降解速率恒定,相位图呈现杏仁形。但实际数据嘈杂,需要先对数据进行平滑处理。
- 动力学参数估计:使用
scv.tl.recover_dynamics函数推断剪接动力学的参数。这些参数包括转录速率(alpha)、剪接速率(beta)和降解速率(gamma)。 - 模型选择:可以选择使用稳态模型(
mode="deterministic")或动态模型(mode="dynamical")。动态模型通过考虑完整的动力学过程,提供更准确的速度估计。
RNA速度计算
- 计算速度向量:使用
scv.tl.velocity函数计算每个细胞的速度向量。速度向量表示细胞在基因表达空间中的运动方向和速度。 - 构建速度图:通过
scv.tl.velocity_graph函数构建速度图,该图描述了细胞之间的转移概率,反映了细胞状态的变化。
可视化
- 速度向量投影:将速度向量投影到低维嵌入空间(如UMAP)中,以便可视化细胞的动态变化。可以使用
scv.pl.velocity_embedding_stream函数绘制流线图。 - 分析细胞状态变化:通过可视化结果,可以观察到细胞状态的迁移方向和动态变化。例如,可以识别出细胞周期的动态变化。
模型验证与分析
- 评估模型拟合度:通过检查相位图和推断出的轨迹,评估模型对数据的拟合程度。
- 识别关键基因:使用
scv.tl.rank_velocity_genes函数识别具有显著速度变化的基因,这些基因可能在细胞状态变化中起重要作用。 - 细胞周期分析:可以使用
scv.tl.score_genes_cell_cycle函数对细胞周期进行评分,以验证RNA速度分析的结果。
结果
UMAP箭头图显示细胞状态迁移方向。
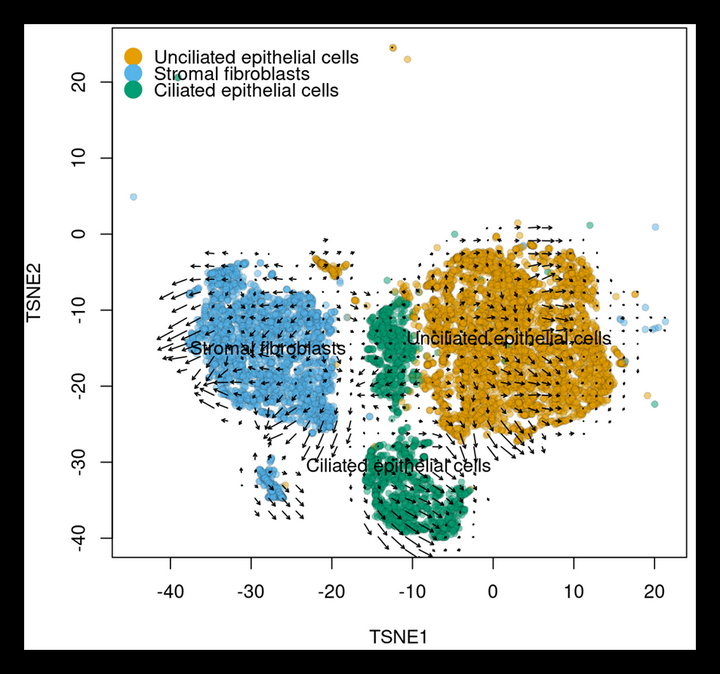 箭头的方向显示了细胞状态转换的动态路径。可以看到,不同细胞类型之间存在一定的转换关系,可能反映了细胞分化、增殖或转分化的过程。某些区域的箭头较为密集,表明这些区域的细胞状态转换较为活跃;某些区域的箭头较为稀疏,表明这些区域的细胞状态转换相对较少
箭头的方向显示了细胞状态转换的动态路径。可以看到,不同细胞类型之间存在一定的转换关系,可能反映了细胞分化、增殖或转分化的过程。某些区域的箭头较为密集,表明这些区域的细胞状态转换较为活跃;某些区域的箭头较为稀疏,表明这些区域的细胞状态转换相对较少
。
差异基因表达分析 Differential gene expression analysis
原因
识别组间(如疾病vs对照)差异表达基因。
差异表达分析旨在判断两组细胞在基因表达模式上的差异是否显著,目的是确定这种差异是否具有统计学意义,而非由随机扰动或噪声引起。
分析有两个假设:
- Null假设(零假设):假设两组细胞的基因表达模式没有显著差异。
- Alternative假设(备择假设):假设两组细胞的基因表达模式存在显著差异。
选择差异表达测试
- 如果数据遵循正态分布,可以使用t检验。
- 如果数据不遵循正态分布,可以考虑使用秩和检验或非参数检验。
- 在更复杂的情况下,可以使用广义线性模型(GLM),根据数据类型(如计数数据)选择合适的噪声模型。
常用的差异表达测试
t检验
假设数据符合正态分布。
秩和检验(Rank Sum Test)
非参数方法,适用于非正态分布数据。
eg
Wilcoxon检验:适合单细胞稀疏数据。
- 工作流程
- 数据准备:将基因表达数据分为两组(例如疾病组和对照组)。
- 秩和计算:将两组数据合并后按表达值从小到大排序,并赋予每个数据点一个秩次(排名)。然后分别计算两组数据的秩次和。
- 统计检验:通过比较两组秩次和的差异,计算出一个p值来判断两组数据是否存在显著差异。
- 工作流程
似然比检验(Likelihood Ratio Test):比较两个不同模型(全模型和简化模型)的拟合度,以判断哪个模型更适合数据。
- 工作流程
- 数据准备:
- 准备两组数据,例如疾病组和对照组的基因表达数据。
- 确定零假设(H₀)和备择假设(H₁)。例如,零假设可能是某个基因在两组之间没有差异表达,而备择假设是该基因存在差异表达。
- 模型拟合:
- 简化模型(Null Model):在零假设下拟合模型,通常假设基因在两组之间没有差异表达。
- 全模型(Alternative Model):在备择假设下拟合模型,允许基因在两组之间存在差异表达。
- 计算似然函数:
- 对于简化模型和全模型,分别计算似然函数(L₀和L₁)。似然函数衡量了模型在给定数据下的拟合优度。
- 计算似然比统计量:
- 计算似然比统计量(λ):λ=L1L0,其中L₀是简化模型的似然值,L₁是全模型的似然值。
- 通常对似然比取对数并乘以2,得到似然比检验统计量:−2ln(λ)。
- 显著性评估:
- 在零假设下,似然比检验统计量服从卡方分布(χ²)。根据卡方分布计算p值,判断模型之间的差异是否显著。
- 数据准备:
- 工作流程
Wall检验:评估回归系数的后验分布是否显著不同于零。
- 工作流程:
- 数据准备:
- 准备数据,包括自变量(如基因表达水平)和因变量(如表型或状态)。
- 模型拟合:
- 拟合一个回归模型(如线性回归或逻辑回归),得到回归系数的估计值(β)。
- 计算标准误:
- 计算回归系数的标准误(SE)。标准误衡量了回归系数估计的不确定性。
- 计算Wald统计量:
- 对于每个回归系数,计算Wald统计量:Z=SE(β)β,其中β是回归系数的估计值,SE(β)是其标准误。
- 显著性评估:
- 在零假设下,Wald统计量服从标准正态分布(N(0,1))。根据标准正态分布计算p值,判断回归系数是否显著不同于零
- 数据准备:
- 工作流程:
差异表达模型
- 广义线性模型(GLM):当数据较复杂时,可以使用广义线性模型。它允许根据数据类型(例如,计数数据)选择不同的噪声模型,如正态分布、泊松分布或负二项分布。
eg - MAST(Model-based Analysis of Single-cell Transcriptomics)
- 特点:专门用于处理单细胞RNA-seq数据中的零膨胀问题。
- 单细胞数据中存在大量零值(即某些基因在许多细胞中不表达),MAST通过零膨胀模型来处理这些零值,从而更准确地识别差异表达基因。
- 工作原理:
- 零膨胀模型:MAST假设数据中存在两种类型的零值:一种是由于技术噪声导致的零值,另一种是基因本身不表达导致的零值。
- 广义线性模型(GLM):MAST使用GLM来建模基因表达数据,考虑了基因表达的变异性。
- 统计检验:通过比较不同组之间的基因表达差异,计算出每个基因的对数2倍变化(log2FC)和p值。
- 特点:专门用于处理单细胞RNA-seq数据中的零膨胀问题。
结果
DEGs(差异表达基因)列表。
基因富集 Gene set enrichment analysis(GSEA)
原因
解释DEGs(差异表达基因)的生物学意义。
- 基因集是预定义的基因组,它们共享某些生物学特性或功能。
基因集富集分析 (Gene Set Enrichment Analysis, GSEA) 旨在通过识别差异表达基因 (DEGs) 集合中过度代表的基因类群或基因集,进而帮助理解这些基因与特定疾病表型的关联。这个方法帮助我们将大量的差异表达基因进行分类和解释。
基因集富集分析对于理解大规模差异表达基因的生物学意义非常有用,但结果可能会受到随机签名的影响,因此要小心解释。
必须考虑基因集的大小和显著性,避免过度解释那些较弱或不具信息性的术语。
基因集富集分析工具
- G Profiler:
- 一个基于网页的工具,可以进行基因集富集分析。
- 也可以通过 Python API 进行访问。
- 用户上传差异表达基因列表,得到富集分析结果。
- 提供来自不同基因集的术语(如 基因本体论 (GO) 术语),帮助分析哪些生物过程或功能在数据中过度代表。
- MetaScape:
- 另一个基于网页的工具,可以进行基因集富集分析。
- 用户上传差异表达基因列表,获得富集分析结果。
工作流程
基因排序
目的:根据基因的表达差异程度对所有基因进行排序,为后续富集分析提供基础。
过程:
- 选择排序指标:通常使用对数2倍变化(log2FC)、P值、t统计量等指标。例如,log2FC表示基因在不同条件下的表达水平变化倍数,能够直观反映基因表达的差异程度。
- 排序操作:根据选定的指标对所有基因进行排序,生成一个有序的基因列表。例如,按照log2FC从高到低排序,高表达差异的基因排在前面。
富集分数计算(Enrichment Score, ES)
目的:评估预先定义的基因集在排序基因列表中的富集程度。
过程:
- 初始化:设置一个运行总和统计量(running sum statistic)为零。
- 遍历基因列表:逐个检查排序后的基因列表,当遇到基因集中的基因时,增加运行总和统计量;遇到不在基因集中的基因时,减少运行总和统计量。
- 计算ES:运行总和统计量在遍历过程中的最大值或最小值即为富集分数(ES)。
- ES反映了基因集在基因列表中的富集程度,正值表示基因集富集在列表的顶部(高表达),负值表示富集在底部(低表达)。
标准化富集分数(Normalized Enrichment Score, NES)
目的:消除基因集大小对富集分数的影响,使不同基因集的富集分数具有可比性。
过程:
- 标准化处理:将每个基因集的ES除以该基因集在随机排列的基因列表中计算得到的ES的平均值。这样可以校正基因集大小对富集分数的影响,得到标准化富集分数(NES)。
- NES的意义:NES的绝对值越大,表示基因集的富集程度越显著。通常,|NES| > 1被认为是显著富集的阈值。
显著性评估
目的:判断观察到的基因集富集是否具有统计学意义。
过程:
- 随机排列:对基因列表进行多次随机排列(如1000次),在每次随机排列的基因列表中重新计算基因集的ES。这样可以生成一个随机背景下的ES分布。
- 计算p值:将观察到的ES与随机排列得到的ES分布进行比较,计算出观察到的ES在随机分布中的位置,从而得到p值。p值越小,表示基因集的富集越显著。
- FDR校正:为了控制多重假设检验中的假阳性率,通常对p值进行FDR(False Discovery Rate)校正。FDR校正后的q值小于某个阈值(如0.25)被认为是显著的。
通过上述步骤,GSEA能够系统地评估预先定义的基因集在排序基因列表中的富集情况,并通过标准化富集分数(NES)和显著性评估(p值和FDR)来判断基因集的富集是否具有统计学意义。
验证富集分析关键点
- 使用互补方法进行验证:如果你认为富集结果很重要,最好使用互补方法验证这些结果,以确认它们确实具有生物学意义。
- 互补方法
- 警惕选择性报告:由于基因集富集分析可能返回多种结果,研究人员可能会倾向于选择那些看起来相关性强的术语,而忽略可能是虚假的阳性或弱关联的术语。因此,在解读结果时要小心这种偏倚。
- 考虑假阳性:研究表明,随机基因表达签名往往会与疾病结果显著相关,例如乳腺癌。
- 研究表明,随机基因集在进行基因集富集分析时,即使它们没有任何生物学相关性,也可能获得显著的结果。 这是富集分析的一个常见陷阱:可能观察到统计学显著性,但结果可能是随机的。
- 假阳性是基因集富集分析中的常见问题,因此应该使用其他方法(例如 分子签名数据库 (MSigDB))进行验证。
- 示例:一个与乳腺癌结果相关的随机基因集,可能在富集分析中返回非常低的 p 值,但这并不意味着它具有生物学意义,可能只是一个假阳性。
结果
这种方法可以帮助研究人员揭示差异表达基因背后的生物学过程或通路。
例如“炎症反应”通路的显著富集(FDR < 0.05)可能表明该通路在研究的生物学过程中发挥了重要作用。
2.染色质可及性(scATAC-seq)
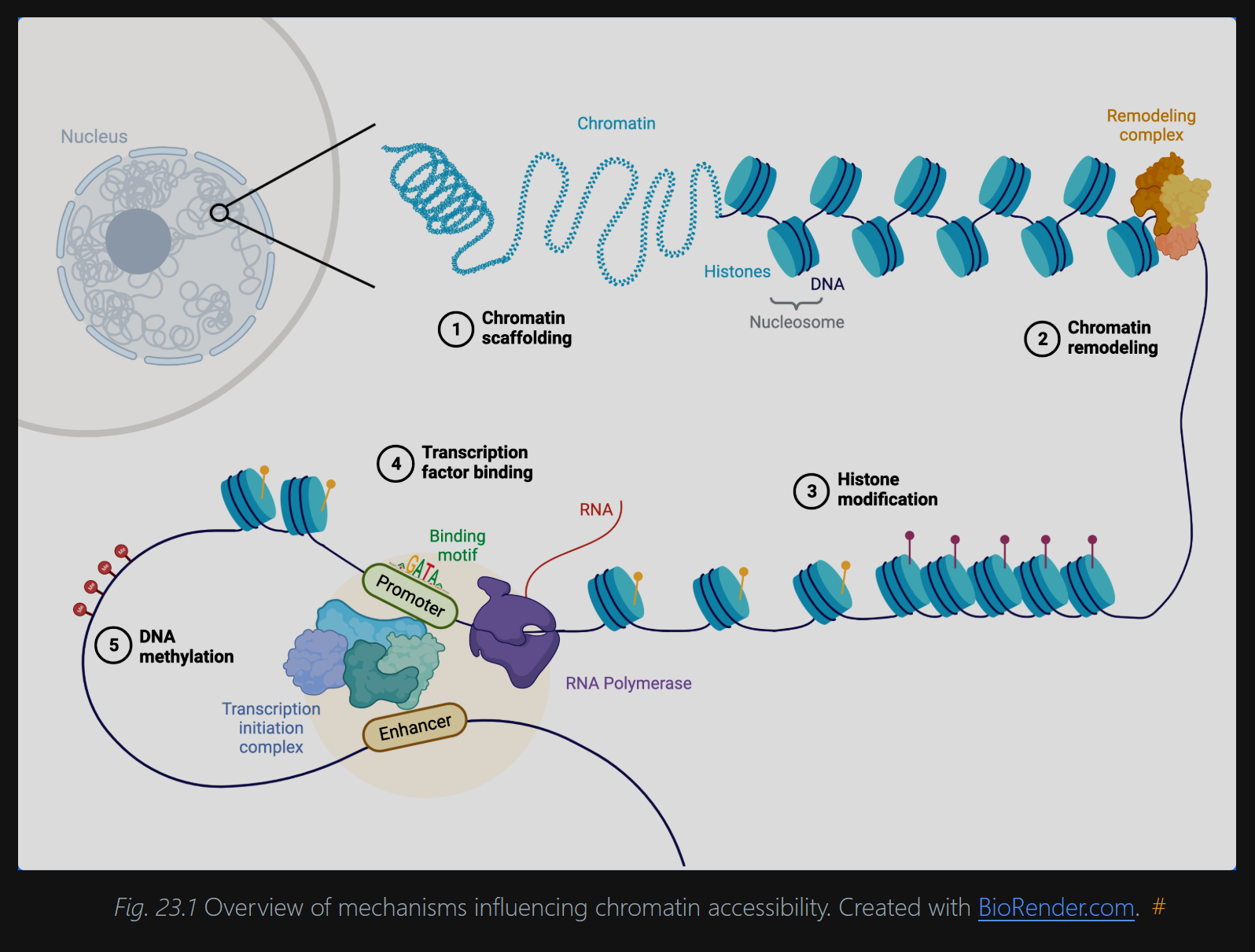
背景
单细胞染色质可及性(scATAC-seq)是一种用于研究单细胞内染色质开放性和基因调控状态的技术。
通过检测染色质开放区域,揭示基因调控状态,补充转录组数据的调控机制信息。
生物体的每个细胞都共享相同的 DNA,具有相同的功能单元集,称为基因。
考虑到这一点,是什么决定了细胞的巨大多样性?
在前面的章节中,我们看到可以从每个细胞中的基因表达谱中推断出细胞的身份和功能。
基因表达的控制是由 DNA 甲基化、组蛋白修饰和转录因子活性等调节机制的复杂相互作用驱动的。染色质可及性在很大程度上反映了细胞的综合调节状态,作为描述细胞身份的 mRNA 水平的正交信息层。
此外,探索染色质可及性谱有助于更深入地了解基因调控机制和细胞分化过程,而 scRNA-seq 数据可能无法捕获这些机制和细胞分化过程。
- 染色质开放性
- 染色质结构的某些区域变得相对松散,使得DNA更容易被细胞内的其他分子(如转录因子)访问。
- 当染色质开放时,特定的基因更容易被转录成RNA,从而被表达。
- 基因调控状态
- 基因调控是指细胞如何控制基因的开启(表达)或关闭(沉默)的过程。
- 基因调控状态涉及到细胞内多种因素,包括转录因子、增强子、沉默子等,它们可以促进或抑制基因的表达。
- 在单细胞水平上,基因调控状态可以揭示细胞特定功能和身份,因为不同细胞类型的基因表达模式是不同的。
染色质的开放程度与基因的激活和调控密切相关。ATAC-seq技术通过测定DNA片段在染色质开放区域的插入情况来评估染色质的可及性,这些区域往往是基因表达的调控位点。
- 应用:scATAC-seq可以揭示哪些基因区域在特定细胞中处于开放状态,并推测哪些转录因子可能在调控基因表达。它有助于理解细胞在不同生理状态下如何调节基因的转录活性,特别是在细胞发育、分化以及应对外界刺激时的基因调控。
- 关键点:ATAC-seq的技术优势在于能够提供基因调控的全景图,但其稀疏数据特性(仅部分基因组区域可被访问)和噪音问题是挑战。
实验流程
细胞核分离与Tn5转座酶处理
目前,市售试剂盒是使用最广泛的实验方案。以10x Multiome检测生成的数据为例(稍作更改也适用于单峰10x单细胞ATAC-seq检测生成的数据):
- 关键原理是Assay for Transposase-Accessible Chromatin with High-Throughput Sequencing(ATAC-seq)。
- 从目标组织的单细胞悬液开始,裂解细胞膜保留完整核(Dounce匀浆器),提取细胞核。
- 使用Tn5转座酶结合染色质开放区域并产生标记化的DNA片段,切割开放染色质区域并添加测序接头。
library构建与测序
- 将细胞核加载到10x Chromium Controller上,形成含有凝胶珠和单细胞的液滴(GEMs),在每个液滴中对RNA分子和DNA片段进行条形码标记。
- 最终获得的scATAC-seq和scRNA-seq文库经过双端测序并与参考基因组比对。
分析流程
数据预处理
原因
scATAC-seq数据高度稀疏(每个细胞仅开放部分区域)且技术噪声高(如低测序深度、核小体污染)。
目标
- 过滤低质量细胞
- 生成细胞×峰/箱矩阵
步骤
质量控制QC
- 方法:
- 使用Python、muon、ArchR工具进行质量控制,计算细胞的测序深度、TSS富集分数等指标。
- 通过双端测序数据的分析,评估细胞的信号与噪声比。
- 过滤:
- 低测序深度细胞(如总片段 <1,000)
- 低TSS富集分数(如TSS得分 <2)
- 高核小体信号(如核小体区域占比 >30%)
- 指标:
- 测序深度(总片段数):反映细胞的测序数据量。
- 测序深度是指测序得到的碱基总量与目标基因组大小的比值,也可以用总片段数来表示,即从一个细胞中获得的测序读数的总数。它反映了对细胞基因组的覆盖程度,是衡量细胞测序数据量的重要指标。测序深度越高,数据的可靠性和分析的准确性通常越高
- TSS富集分数:评估开放区域信号强度,用于判断实验质量。
- TSS富集分数是指以转录起始位点(TSS,转录起始位点)为中心的片段与TSS侧翼区域中的片段的比率。在ATAC-seq实验中,TSS区域通常是无核小体的,因此转座酶更容易切割,从而在TSS区域产生更多的短片段。TSS富集分数越高,说明实验中TSS区域的开放性越好,实验质量越高
- 核小体信号:通过开放区域长度评估核小体污染程度
- 核小体信号是通过分析DNA片段长度来评估核小体污染程度的指标。在ATAC-seq中,无核小体区域(NFR)的片段长度通常小于100 bp,而核小体单体的片段长度约为200 bp。核小体信号通过计算单核小体与无核小体片段的近似比率来量化。较高的核小体信号值可能表示核小体污染较严重,影响染色质开放区域的准确检测。
- 测序深度(总片段数):反映细胞的测序数据量。
二值化
- 数据特征:scATAC-seq数据具有高度稀疏性,因为二倍体生物的每个细胞中只有两个DNA拷贝,导致许多特征的计数为零。
- 为应对稀疏性,一些方法采用二值化,即将特征的可及性简化为“有”或“无”,但这种方法可能导致信息丢失。
峰检测与矩阵生成
- 峰检测:
- 峰检测是识别基因组中开放区域或富集区域的过程
- 开放区域是指基因组中染色质结构较为松散、对核酸酶敏感的区域。
- 富集区域是指基因组中某些特定蛋白质(如转录因子、组蛋白修饰等)结合较为集中的区域。
- MACS2识别全样本开放区域,合并为统一峰集
- 峰检测是识别基因组中开放区域或富集区域的过程
- 矩阵构建:
- 细胞×峰矩阵:统计每个细胞在各峰的片段数(稀疏矩阵),从而生成一个矩阵。矩阵的行通常代表不同的细胞,列代表基因组的不同窗口。
- 细胞×箱矩阵:将基因组划分为5 kb窗口(箱),统计窗口内片段数 。从而生成一个矩阵。矩阵的行通常代表不同的细胞,列代表基因组的不同窗口。
结果
- 过滤后的细胞×峰矩阵(如保留10,000个峰和5,000个细胞)
- 稀疏性降低(如非零条目占比从0.1%提升至1%)
降维与注释
原因
高维稀疏矩阵难以直接分析,需压缩维度并关联生物学意义。
目标
- 降维至低维空间(如20维)
- 注释细胞类型
步骤
降维
- LSI(潜在语义索引):TF-IDF加权 → SVD分解 → 提取Top LSI成分
- LSI是一种基于文本摘要的降维技术,其核心是通过SVD分解将高维稀疏矩阵分解为低维矩阵。
- 在生物信息学中,首先对基因表达或染色质可及性数据进行TF-IDF加权,以突出重要特征并减少常见特征的影响,然后通过SVD分解提取Top LSI成分,从而将数据压缩到低维空间(如20维)。
- 因为数据高度稀疏、非线性,以及出于生物学关联与计算效率等考虑,此处选择LSI方法进行降维。
- 批次校正:Harmony对齐不同批次的LSI嵌入
- Harmony是一种用于校正批次效应的算法,通过将不同批次的LSI嵌入对齐,消除批次间差异,使得后续分析更加准确。
基因活性评分
- Cicero:一款基于R的单细胞染色质可及性实验可视化工具。它通过分析染色质开放性数据,基于共可及性(邻近开放区域共现)来计算基因启动子活性。
- 公式:$A_g = \sum_{p \in P_g} \text{co-accessibility}(p, g) \cdot \text{reads}(p)$
- 其中Ag表示基因g的启动子活性,Pg是与基因g启动子邻近的开放区域,co-accessibility(p,g)表示区域p与基因g的共可及性,reads(p)表示区域p的测序读数。
- 公式:$A_g = \sum_{p \in P_g} \text{co-accessibility}(p, g) \cdot \text{reads}(p)$
聚类与注释
- 聚类:Leiden算法基于LSI嵌入的KNN图分区
- 注释:
- 差异可及性区域(DARs):Wilcoxon检验筛选簇特异性开放峰
- 这些区域在特定细胞簇中表现出显著的染色质开放性差异,从而确定细胞类型的特征区域
- 基因活性:匹配启动子活性与已知细胞类型标记基因
- 根据标记基因的表达模式对细胞簇进行注释。例如,簇1包含CD4基因座的DARs,则可注释为CD4+ T细胞
- 差异可及性区域(DARs):Wilcoxon检验筛选簇特异性开放峰
结果
- UMAP图显示按类型聚集的细胞(如T细胞、B细胞、单核细胞)
- 注释标签(如簇1→CD4+ T细胞,DARs包含CD4基因座)
细胞调控分析
原因
开放染色质区域提示调控元件(如增强子)活性,需解析调控网络。
目标
- 识别关键转录因子(TF)
- 推断基因调控网络
方法
TF活性分析
TF指的是转录因子(Transcription Factor)。转录因子是一类能够与DNA特异性结合的蛋白质,它们通过与基因启动子区域或其他调控元件结合,调控基因的转录过程
工具:
- chromVAR:计算每个细胞的TF motif可及性偏差,从而评估TF活性
- TF活性是指转录因子在细胞中调控基因表达的能力,通常通过其结合位点的染色质可及性或靶基因表达水平来推断。
- TF motif可及性偏差是指在单个细胞中,特定转录因子结合位点(motif)的染色质可及性与整体预期可及性之间的差异。
- 公式:$Z_{TF} = \frac{\text{Observed Motif Accessbility} - \text{Expected}}{\text{Standard Deviation}}$
- Observed Motif Accessibility表示实际观测到的motif可及性,Expected是基于背景模型的预期可及性,Standard Deviation是标准差。
Footprinting分析
- TOBIAS:检测TF结合位点的开放性下降(因TF占据阻碍Tn5切割)的工具
- 通过分析ATAC-seq数据,识别出因TF结合而阻碍Tn5切割的区域,从而推断出TF的结合位点。帮助研究者了解TF在基因调控中的作用。
结果
- TF活性矩阵(如PU.1在单核细胞中高活性)
- 调控网络图(如SPI1→IRF8→CEBPA级联调控髓系分化)
3. 表面蛋白(CITE-seq)
背景
表面蛋白分析(CITE-seq)是一种结合单细胞转录组学与表面蛋白表达分析的方法,提供多维度细胞表型信息。
CITE-seq通过使用抗体衍生标签(ADT,Antibody-Derived Tags)标记细胞表面的蛋白质,然后与传统的RNA测序一起进行分析,提供了细胞转录组和表面蛋白的联合信息。
抗体衍生标签(ADT)
- 一种利用抗体来识别和标记细胞表面特定蛋白的技术。在这种技术中,抗体被设计成带有可以被测序检测的特殊DNA序列(即标签)。当这些带有标签的抗体与细胞表面的蛋白结合时,这些标签也就间接地标记了相应的蛋白。通过测序这些DNA标签,研究人员可以确定细胞表面蛋白的存在和相对丰度。
单细胞RNA-seq数据只能反映转录组信息,而CITE-seq技术可以同时捕获细胞表面蛋白表达丰度,为研究细胞过程提供更全面的信息。
CITE-seq数据包含两种模态:基因表达数据和抗体衍生标签(ADT)数据,二者分布不同,需要不同的预处理方法。
ADT数据可以用于识别细胞类型、检测双细胞(doublets)等,但其数据分布更复杂,背景噪声更高。
应用:CITE-seq技术能够在单细胞水平上同时分析细胞的转录信息和表面蛋白的表达情况,帮助研究细胞亚群的特征、免疫细胞的状态(例如T细胞的激活与免疫反应)、以及细胞间的相互作用等。
关键点:CITE-seq的优势是能够同时获取细胞的转录组和表面蛋白信息,但数据质量受到抗体特异性、背景噪音和抗体反应的影响。
实验流程
抗体标记:寡核苷酸偶联抗体(TotalSeq-B)标记细胞表面蛋白。
联合测序:10x Multiome同时捕获RNA与抗体衍生标签(ADT)。
分析流程
ADT标准化
原因
ADT数据受背景噪音(非特异抗体结合)和文库大小影响,需标准化。
目标
- 去除技术噪声
- 校正文库大小差异
步骤
背景校正 DSB(Denoised and Scaled by Background)
- 目的:去除ADT数据中的背景噪音,这些噪音可能来源于非特异性抗体结合
- 方法
- 使用同型对照抗体(无目标蛋白)估计背景分布,然后通过以下公式对原始ADT数据进行校正
- 公式:$ADT_{corrected} = \frac{ADT_{raw} - \mu_{background}}{\sigma_{background}}$
- 其中$μ_{background}$和$σ_{background}$分别表示背景分布的均值和标准差
标准化 CLR(Centered Log Ratio)
- 目的:消除文库大小差异对ADT数据的影响
- 方法
- 通过计算每个细胞中每个ADT特征的中心对数比率进行标准化
- 公式:$CLR(ADT_i) = \ln\left(\frac{ADT_i}{\text{geometric mean}(ADT)}\right)$
- 其中geometric mean(ADT)是所有ADT特征的几何平均值
- 公式:$CLR(ADT_i) = \ln\left(\frac{ADT_i}{\text{geometric mean}(ADT)}\right)$
结果
- 校正后的ADT矩阵(背景信号趋近于零)
多模态整合
原因
RNA与ADT数据分布不同,需联合建模以揭示一致细胞状态。
目标
- 生成共享潜在空间
- 提升细胞分群分辨率
方法
TotalVI(Variational Inference)
- 模型:变分自编码器联合建模RNA(负二项分布)与ADT(高斯分布)
- 输入:
- RNA表达矩阵 X:包含 N 个细胞和 G 个基因的UMI计数。
- ADT表达矩阵 Y:包含 N 个细胞和 T 个蛋白质的UMI计数 - 模型构建
- 假设分布:
- RNA表达量服从负二项分布,用于处理RNA-seq数据中的过离散性。
- ADT数据服从高斯分布,用于处理连续的蛋白质表达数据。
- 生成过程:
- 对于每个细胞 n,生成潜在变量 $z_n$,并计算RNA和蛋白质的表达参数。
- RNA表达量 $x_{ng}$服从负二项分布,蛋白质表达量 $y_{nt}$ 服从负二项混合分布。
- 假设分布:
- 推断过程
- 使用变分推断(Variational Inference)和自动编码变分贝叶斯(Auto-Encoding Variational Bayes)来学习模型参数。
- 通过优化近似后验分布 $q_η(z_n∣x_n,y_n,s_n)$ 来估计潜在变量
- 输出:
- 共享潜在变量:生成一个低维的共享潜在空间(如30维),用于后续的细胞分群和注释。
- 去噪表达值:提供去噪后的RNA和ADT表达值。
- 蛋白背景概率:估计每个细胞中每个ADT特征的背景概率
跨模态聚类
- 目的:基于RNA和ADT数据的相似性,构建加权细胞图,以提高细胞分群的分辨率
- 方法:
- 加权最近邻(WNN):通过计算RNA和ADT数据的相似性,构建加权的最近邻图(KNN)。RNA和ADT的权重默认分别为0.5。有对应的python包。
- 参数:可以根据数据特点调整RNA和ADT的权重,以优化聚类结果。
- 下游分析:基于WNN图进行UMAP降维和细胞分群,最终得到更准确的细胞类型分群结果
结果
- UMAP图中细胞按类型聚集(如CD4+ T细胞、CD8+ T细胞、NK细胞)
- 蛋白标记验证(如CD3ε在T细胞中高表达)
4.适性免疫受体库(AIRR)
背景
免疫受体库分析揭示B/T细胞的克隆多样性及其抗原特异性。
概念
免疫受体 IR
免疫受体(Immune Receptors, IR)是识别潜在有害抗原和毒素的关键结构。它们通常位于特定免疫细胞的膜上,能够识别有害物质并启动保护机制。根据识别的抗原类型和细胞类型,IR可以分为以下几类:
- 模式识别受体(Pattern Recognition Receptors, PRRs):识别病原体相关分子模式(PAMPs)。
- 杀伤激活/抑制受体(Killer Activator/Inhibitor Receptors, KARs/KIRs):识别宿主细胞的异常。
- 补体受体(Complement Receptors):与补体蛋白结合。
- Fc受体(Fc Receptors):与抗体-表位复合物结合。
- 细胞因子受体(Cytokine Receptors):与细胞因子结合。
- B细胞受体(B-cell Receptors, BCRs):识别可溶性或膜结合的表位。
- T细胞受体(T-cell Receptors, TCRs):与MHC分子结合的线性表位。
适性免疫受体 AIR
适应性免疫受体(Adaptive Immune Receptors, AIR)主要由B细胞和T细胞产生,通过V(D)J重组机制形成高度多态性的受体序列。V(D)J重组通过以下方式引入序列多样性:
- 组合多样性(Combinatorial Diversity):不同V、D、J基因的组合。
- 连接多样性(Junctional Diversity):基因接口处插入核苷酸。
- 体细胞高频突变(Somatic Hypermutation):在BCR结合位点发生突变,用于亲和力成熟。
AIR的三个高变区(Complementary Determining Regions, CDRs)1-3是与目标抗原结合的关键区域,其中CDR3跨越V、D、J基因的交界处,是AIR中最具多样性的部分。
免疫受体测序(VDJ-sequencing)
VDJ测序(VDJ-sequencing)是一种高通量测序技术,专门用于分析B细胞受体(BCR)和T细胞受体(TCR)的基因重排多样性。其核心目标是解析免疫系统中B细胞和T细胞受体基因的V(Variable)、D(Diversity)、J(Joining)基因片段在发育过程中的重组模式,从而揭示适应性免疫系统的多样性特征、克隆演化规律或疾病相关的异常免疫应答。
VDJ测序通过单细胞RNA测序重建V(D)J链序列,用于分析AIR的多样性和功能。常见的单细胞分离方法包括:
- 荧光激活细胞分选(Fluorescence Activated Cell Sorting, FACS):基于荧光标记的细胞分选。
- 磁激活细胞分选(Magnetic-Activated Cell Sorting, MACS):利用磁珠标记目标细胞。
- 激光捕获显微切割(Laser Capture Microdissection, LCM):从显微镜样本中提取目标细胞。
- 微流控技术(Microfluidics):处理微量细胞悬液。
AIRR
免疫受体库分析(AIRR,Adaptive Immune Receptor Repertoire sequencing)专注于分析免疫系统中T细胞受体(TCR)和B细胞受体(BCR)的多样性。
这些受体是免疫细胞识别并与抗原(如病原)相互作用的关键分子。
- 应用:通过测序TCR和BCR的V(D)J基因重排,研究人员可以分析免疫系统的多样性和功能性。AIRR技术可以帮助研究免疫反应的动态变化,如抗体和T细胞的特异性扩增,进而在疫苗开发、免疫治疗以及自身免疫病研究中提供重要数据。
- 关键点:免疫受体库的挑战在于如何识别和分析大量的TCR/BCR序列及其功能,特别是在免疫系统遭遇不同病原或治疗干预时,如何准确分析受体的变化。
分析流程
数据预处理
- 序列重建:使用MiXCR、TRUST4等工具重建TCR/BCR序列。
- 细胞分离与标记:使用FACS、MACS等方法分离目标细胞。
- IR信息对齐:将IR信息从单细胞水平对齐到细胞条码上。
- 质量控制:去除低质量序列,标记双细胞。
克隆型分析与特异性预测
- 克隆型分析:通过V基因和CDR3序列识别克隆型,分析克隆型的组成和多样性。
- 克隆型定义:通过比较V(D)J CDR3序列的相似性来定义克隆型。
- 特异性预测:使用数据库查询(如IEDB、vdjDB)或机器学习工具(如ERGO-II)预测TCR/BCR的抗原特异性。
- 克隆型分析革新:
- BCR进化树:IgPhyML(最大似然法)解析体细胞超突变路径;
- TCR特异性预测:TCRdist3(图神经网络)跨HLA亚型泛化能力提升。
- 多组学关联:
- 转录-受体协同:TESSA(多视角自编码器)识别克隆扩增相关通路(如IFN-γ信号)。
克隆型分析
通过V基因和CDR3序列识别克隆型,分析克隆型的组成和多样性。
克隆型定义
- BCR克隆型:相同VH-JH和VL-JL链,CDR3氨基酸序列相似度>85%。
- TCR克隆型:相同TRA-TRB链,CDR3氨基酸序列完全相同。
克隆多样性指标
- Shannon指数:用于量化克隆丰富度。
- 克隆扩增度:例如肿瘤浸润T细胞中最大克隆占比可能>20%。
多组学整合
- TESSA工具:联合转录组与TCR序列,识别克隆相关基因模块。例如,克隆扩增的T细胞可能高表达PD-1等免疫检查点基因。
结果输出
- 克隆型统计表:例如前10大克隆占比50%。
- 克隆-功能热图:展示不同克隆的功能相关性,如克隆A高表达细胞毒性基因。
抗原特异性预测
特异性分析旨在确定B细胞和T细胞的AIR序列与特定抗原的结合能力。
方法
数据库匹配
- VDJdb数据库:查询已知TCR-抗原对,例如CMV特异性TCR。
- IEDB数据库:检索BCR表位信息,如流感HA蛋白。
机器学习预测
- ERGO-II工具:基于TCR序列和HLA类型,通过深度学习模型预测抗原,输出概率评分。例如,TCR-X对EBV抗原的预测概率为0.92。
结果输出
- 抗原特异性注释:例如克隆Y靶向HPV E6蛋白。
- 潜在新抗原列表:为肿瘤新生抗原等研究提供候选。
多组学整合
将AIR信息与转录组学数据整合,可以更全面地分析免疫细胞的功能和状态。
整合方法
- TESSA:通过贝叶斯模型将TCR序列和转录组数据嵌入到共享低维空间中。
- CoNGA:基于图论方法,通过GEX和TCR的相似性构建细胞克隆型网络。
- mvTCR:使用深度变分自编码器将GEX和TCR数据压缩到共享低维表示中。
- Benisse:针对B细胞,将BCR序列嵌入数值空间并与GEX数据结合。
应用案例
- NeoDisc工具:整合基因组、转录组和质谱数据,预测HLA结合的肽段,结合自体T细胞反应进行验证,能够系统地优先排序肿瘤特异性抗原
5. 空间转录组
背景
空间转录组保留细胞原位位置信息,解析组织微环境。
- 传统单细胞技术的局限性:传统单细胞测序技术虽然能够解析细胞身份及其基因组依赖关系,但在分析过程中会丢失细胞的空间信息。而空间信息对于许多生物学问题至关重要。
- 空间解析基因组学的出现:空间解析基因组学技术通过在测量基因组规模的组学数据的同时保留空间信息,解决了这一问题。这些技术能够测量转录组、蛋白质组
实验流程
组织切片与捕获:Visium芯片(10x Genomics)固定组织切片并释放mRNA至空间条形码斑点。
探针杂交:Slide-seqV2使用DNA纳米球阵列捕获空间坐标。
分析流程
空间解卷积
原因
Visium等技术的每个斑点含多个细胞,需解析细胞类型组成。
斑点(spot)是指芯片上用于捕获mRNA的微小区域。
每个Visium芯片包含多个捕获区,每个捕获区由大约5000个斑点组成。
这些斑点的直径为55微米,相邻斑点的圆心距为100微米。每个斑点上分布有数百万个含有poly(dT)的寡核苷酸链,这些链可以捕获细胞释放的mRNA。
由于斑点的直径相对较大,每个斑点通常可以捕获多个细胞的mRNA,具体数量取决于细胞的大小和组织类型。例如,在人类大脑皮层组织中,每个斑点平均可以捕获3.3个细胞。
因此,在分析Visium数据时,需要通过空间解卷积等方法来解析每个斑点中不同细胞类型的组成。
目标
- 推断斑点内细胞类型比例
- 定位细胞类型至空间坐标
步骤
参考构建
使用单细胞参考,即通过单细胞RNA测序(scRNA-seq)注释的细胞类型表达谱
- 单细胞参考:scRNA-seq注释的细胞类型表达谱
反卷积模型
- Cell2Location:是一种贝叶斯模型,它将斑点表达拟合为细胞类型比例的线性组合
- 公式:$Y_{spot} = \sum_{c} w_c \cdot X_c + \epsilon$
- $Y_{spot}$表示斑点的表达量,$w_c$是细胞类型的比例,$X_c$是细胞类型的表达谱,ϵ是误差项
- 公式:$Y_{spot} = \sum_{c} w_c \cdot X_c + \epsilon$
结果
空间映射图(如肿瘤区域富集T细胞和巨噬细胞)
空间模式分析
原因
基因表达的空间变异反映组织功能分区(如肿瘤边界炎症梯度)。
方法
空间可变基因(SVGs)
- SpatialDE:
- 原理:通过高斯过程回归检测基因表达的空间自相关性,是一种用于识别空间变异基因的方法。
- 特点:具有无监督、非参数和非线性表达模式识别等特点,能够自动基于空间共表达的基因分组。
- 结果:SVGs列表,例如胶原蛋白基因在纤维区域高表达。
空间邻域网络
- Giotto:
- 原理:基于空间坐标构建Delaunay三角网,计算邻域共现细胞类型。
- 结果:细胞类型互作网络(如癌细胞与成纤维细胞显著共定位)
结果
- 空间热图(如IFN-γ信号在肿瘤-基质交界处活跃)
- 空间驱动通路(如缺氧通路在肿瘤核心富集)
6. 多组学整合
背景
多组学整合揭示不同分子层(如RNA、ATAC、蛋白)的协同调控。
近年来,出现了几种技术,使我们能够在单个单元中测量多种模态。在这种情况下,模态是指我们可以在每个单元格中捕获的不同类型的信息。例如,CITE-seq 允许测量同一细胞中的基因表达和表面蛋白丰度。或者,使用 Multiome 检测等配对 RNA-seq/ATAC-seq 实验,同时捕获基因表达和染色质可及性。
数据整合方法
| 方法 | 核心原理 | 适用场景 | 优势 | 局限 |
|---|---|---|---|---|
| MOFA+ | 因子分析(概率模型) | 低维潜在空间发现 | 可解释性高 | 对批次效应敏感 |
| WNN | 加权近邻图 | 跨模态聚类 | 计算效率高 | 依赖初始降维质量 |
| GLUE | 变分自编码器+知识图谱 | 非配对数据 | 整合先验生物学知识 | 训练复杂度高 |
| SCOT | 最优传输 | 跨物种对齐 | 无需特征对齐 | 对高维数据计算成本大 |
联合测量数据整合
适用场合
同一细胞的多种模态数据需联合分析以全面解析细胞状态。
方法
MOFA+(Multi-Omics Factor Analysis)
- 模型:概率因子模型分解多组学数据为共享潜在因子
- 输出:因子载荷(如因子1关联RNA中的细胞周期基因和ATAC中的E2F motif)
WNN(Weighted Nearest Neighbor)
- 步骤:
- 单模态降维(RNA→PCA, ATAC→LSI)
- 计算跨模态相似性权重
- 构建加权KNN图进行聚类
结果
混合模态分群(如干细胞群在RNA和ATAC中均显著)
非联合数据整合
适用场合
不同实验的单模态数据集需对齐以进行比较(如跨物种、跨平台)。
方法
SCOT(Single-Cell Optimal Transport)
- 原理:最优传输理论最小化模态间分布差异
- 步骤:
- 单模态降维
- 计算源与目标模态的耦合矩阵
- 投影至公共空间
GLUE(Graph-Linked Unified Embedding)
- 模型:变分自编码器+调控知识图谱引导整合
- 应用:RNA与ATAC联合推断基因-增强子关联
结果
- 对齐的UMAP图(如小鼠与人类皮层细胞跨物种对齐)
- 调控轴(如TF-X调控基因Y通过增强子Z)
查询到参考映射
- 多模态参考映射:使用Multigrate、Bridge等工具将单模态或多模态查询数据映射到多模态参考数据中。
应用案例
- 肿瘤微环境解析:10x Multiome(RNA+ATAC)联合Signac,识别恶性细胞克隆特异性增强子;
- 发育动力学:scRNA-seq + 空间组 + 谱系追踪,重构胚胎器官发生轨迹。
三、Benchmarks
背景
研究目标
- 评估现有方法的性能:通过标准化的测试任务,评估不同数据整合方法在去除批次效应和保留生物变异方面的能力。
- 提供方法选择的指南:为研究人员提供选择合适数据整合方法的参考,特别是在处理复杂数据集时。
- 推动方法开发:通过揭示现有方法的不足,为开发更高效的数据整合方法提供方向。
基准测试的挑战
- 数据集的复杂性:单细胞数据集通常包含多个样本、多种条件和多个实验室的数据,导致复杂的批次效应。
- 方法的多样性:不同的数据整合方法在算法设计、输出格式和预处理要求上存在差异。
- 评估指标的选择:需要平衡批次效应去除和生物变异保留的评估指标。
常用数据集
真实数据集
单细胞RNA-seq数据集
| 数据集名称 | 出处 | 内容及介绍 | 数据集格式 |
|---|---|---|---|
| 人类细胞图谱(Human Cell Atlas, HCA) | HCA官网 | 包含多种组织的单细胞RNA-seq数据,涵盖健康和疾病状态下的细胞类型和基因表达模式。 | 原始数据为FASTQ格式,经过处理后提供矩阵格式(如10X Genomics格式)和注释文件(如H5AD格式)。 |
| 10X Genomics公共数据集 | 10X Genomics官网 | 提供多种单细胞RNA-seq数据集,如PBMC(外周血单核细胞)数据集,广泛用于基准测试和细胞类型注释。 | 原始数据为FASTQ格式,经过处理后提供矩阵格式(如10X Genomics格式)和注释文件(如H5AD格式)。 |
| Tabula Muris | Tabula Muris官网 | 包含小鼠多个器官的单细胞RNA-seq数据,涵盖近100,000个细胞,用于研究细胞类型和基因表达。 | 原始数据为FASTQ格式,经过处理后提供矩阵格式(如10X Genomics格式)和注释文件(如H5AD格式)。 |
| Allen Brain Atlas | Allen Brain Atlas官网 | 提供脑组织的单细胞RNA-seq数据,专注于神经科学和脑功能研究。 | 原始数据为FASTQ格式,经过处理后提供矩阵格式(如10X Genomics格式)和注释文件(如H5AD格式)。 |
| SCPortalen数据库 | SCPortalen官网 | 涵盖人类和小鼠单细胞转录组学数据集,提供单细胞图像访问权限。 | 原始数据为FASTQ格式,经过处理后提供矩阵格式(如10X Genomics格式)和注释文件(如H5AD格式)。 |
| scRNASeqDB数据库 | scRNASeqDB官网 | 包含36个人类单细胞基因表达数据集,涉及174个细胞组的8910个细胞,提供基因表达分析工具。 | 原始数据为FASTQ格式,经过处理后提供矩阵格式(如10X Genomics格式)和注释文件(如H5AD格式)。 |
| CellCommuNet | CellCommuNet官网| | 收集了来自人类和小鼠多种组织在正常和疾病状态下的单细胞RNA测序样本,共376个数据集,4,327,804个单细胞样本,覆盖82种疾病和35种组织类型,包含397种非冗余的细胞类型。 | 原始数据为FASTQ格式,经过处理后提供矩阵格式(如10X Genomics格式)和注释文件(如H5AD格式)。 |
| BloodSpot数据库 | BloodSpot官网 | 提供健康和恶性造血中基因和基因特征的基因表达谱数据库,包含人类和小鼠的数据,专注于血液系统| | 原始数据为FASTQ格式,经过处理后提供矩阵格式(如10X Genomics格式)和注释文件(如H5AD格式)。 |
单细胞ATAC-seq数据集
| 数据集名称 | 出处 | 内容及介绍 | 数据集格式 |
|---|---|---|---|
| 人类外周血单核细胞(PBMCs)单细胞ATAC-seq数据集 | 10x Genomics官网 | 由10x Genomics公司提供,包含8728个细胞的染色质可及性数据,可用于分析人类外周血单核细胞的基因组调控特征。 | 原始数据为FASTQ格式,经过处理后提供BAM格式的比对结果和peak calling结果(如BED格式)。 |
| 人类早期胚胎染色质可及性图谱 | 相关研究 | 通过ATAC-seq技术绘制的人类早期胚胎染色质可及性图谱,揭示了胚胎发育过程中的表观遗传调控机制。 | 原始数据为FASTQ格式,经过处理后提供BAM格式的比对结果和peak calling结果(如BED格式)。 |
| T细胞淋巴瘤(CTCL)ATAC-seq数据集 | 相关研究 | 分析了111个人皮肤T细胞淋巴瘤患者和对照样本的染色质可及性,揭示了T细胞淋巴瘤的表观遗传调控机制。 | 原始数据为FASTQ格式,经过处理后提供BAM格式的比对结果和peak calling结果(如BED格式)。 |
| 人类免疫细胞单细胞ATAC-seq数据集 | 相关研究 | 包含人类免疫细胞的单细胞ATAC-seq数据,用于研究免疫细胞的发育和功能调控。 | 原始数据为FASTQ格式,经过处理后提供BAM格式的比对结果和peak calling结果(如BED格式)。 |
| TCGA ATAC-seq数据集 | TCGA官网 | 提供多种癌症的ATAC-seq数据,涵盖染色质可及性信息,可用于癌症表观遗传学研究。 | 原始数据为FASTQ格式,经过处理后提供BAM格式的比对结果和peak calling结果(如BED格式)。 |
表面蛋白CITE-seq数据集
| 数据集名称 | 出处 | 内容及介绍 | 数据集格式 |
|---|---|---|---|
| CSPA数据库 | CSPA官网 | 提供人类细胞表面蛋白(surfaceome)信息,包含2,886个预测为人类细胞表面蛋白的条目,预测结合位点并生成高质量的结合种子。 | 数据以矩阵格式和注释文件提供,支持多种生物医学研究需求。 |
| SPDB数据库 | SPDB官网 | 单细胞蛋白质组数据库,涵盖133个基于抗体的单细胞蛋白质组数据集,涉及超过3亿个细胞和800多个标记/表面蛋白。 | 数据以矩阵格式和注释文件提供,支持蛋白质组学分析。 |
适性免疫受体库(Immune Repertoire)
| 数据集名称 | 出处 | 内容及介绍 | 数据集格式 |
|---|---|---|---|
| 免疫组库分析数据集 | immunarch包 | 提供12个样本的TCR或BCR注释信息,支持多种免疫组库数据格式,包括immunoseq、mixcr、vdjtools等。 | 数据以表格式存储,支持多种免疫组库分析工具。 |
空间转录组(Spatial Transcriptomics)
| 数据集名称 | 出处 | 内容及介绍 | 数据集格式 |
|---|---|---|---|
| STOmicsDB数据库 | STOmicsDB官网 | 空间转录组数据库,收录了7339篇空间转录组相关文献、228个空转项目数据,涵盖17个物种、128种组织类型,总计7177个样本。 | 数据中心模块提供多种格式数据,包括矩阵格式、注释文件(如H5AD格式)和图像数据。 |
| STimage-1K4M数据集 | STimage-1K4M官网 | 空间转录组学数据集,包含1,149张空间转录组学切片,总计超过400万个具有配对基因表达数据的点。 | 数据集包含注释、元数据、空间坐标、基因表达和图像数据,支持多种分析任务。 |
多组学整合(Multi-Omics Integration)
| 数据集名称 | 出处 | 内容及介绍 | 数据集格式 |
|---|---|---|---|
| ENCODE数据集 | ENCODE官网 | 提供人类基因组的功能注释数据,涵盖多种细胞类型和生物学条件,包含RNA-seq、ATAC-seq等多种组学数据。 | 数据以多种格式提供,包括FASTQ、BAM、BED等格式,支持多组学整合分析。 |
| 高歌课题组单细胞多组学数据集 | 高歌课题组官网 | 提供单细胞多组学数据集,涵盖转录组、染色质可及性等多种组学数据,支持调控推断和多组学整合分析。 | 数据以矩阵格式和注释文件提供,支持多种分析工具。 |
模拟数据集
| 数据集名称 | 出处 | 内容及介绍 | 数据集格式 |
|---|---|---|---|
| Splatter | Splatter官网 | 基于负二项分布生成模拟数据,可控制技术噪声和生物学差异,适用于单细胞RNA-seq数据分析方法的基准测试。 | 模拟数据以矩阵格式(如H5AD格式)提供,包含基因表达矩阵和细胞注释信息。 |
| scDesign2 | 相关研究 | 支持复杂细胞轨迹和批次效应的模拟,适用于单细胞测序数据分析方法的开发。 | 模拟数据以矩阵格式(如H5AD格式)提供,包含基因表达矩阵和细胞注释信息。 |
| SymSim | 相关研究 | 模拟基因表达噪声和细胞分化轨迹,适用于研究细胞分化过程中的基因表达动态。 | 模拟数据以矩阵格式(如H5AD格式)提供,包含基因表达矩阵和细胞注释信息。 |
这些数据集和工具为单细胞测序研究提供了丰富的资源,可用于分析不同组织、物种和实验条件下的基因表达和染色质可及性。
评估指标
单细胞RNA测序(scRNA-seq)的评估指标和框架是确保数据质量和分析可靠性的重要工具。以下是一些常用的评估指标和框架:
均值绝对误差(MAE)
- 用于衡量去噪数据与真实数据之间的差异。MAE值越低,表示去噪后的数据与真实数据越接近。
- 公式为:$$MAE = \frac{1}{n}\sum _{i=1}^{n}|y_{i} - \hat{y}_{i}|$$ 其中,$n$ 是观测值总数,$y_{i}$ 是真实值,$\hat{y}_{i}$ 是预测值。
皮尔逊相关系数(PCC)
- 用于衡量去噪数据与真实数据之间的线性相关性。PCC值在-1到1之间,1表示完全正相关,-1表示完全负相关,0表示无线性相关。
- 公式为:$$r = \frac{\sum _{i=1}^{n} (x_i - \bar{x})(y_i - \bar{y})}{\sqrt{\sum _{i=1}^{n} (x_i - \bar{x})^2} \sqrt{\sum _{i=1}^{n} (y_i - \bar{y})^2}}$$ 其中,$x_{i}$ 和 $y_{i}$ 分别是两个变量的观测值,$\bar{x}$ 和 $\bar{y}$ 分别是它们的均值。
均方误差(MSE)
- 定义:衡量去噪数据与真实数据之间的差异。MSE值越低,表示去噪后的数据与真实数据越接近。
- 公式:$$MSE = \frac{1}{n}\sum _{i=1}^{n}(y_{i} - \hat{y}_{i})^2$$ 其中,$n$ 是观测值总数,$y_{i}$ 是真实值,$\hat{y}_{i}$ 是预测值。
- 取值范围:MSE 的取值范围是非负数。MSE 越小,说明模型的预测值与真实值之间的差异越小,模型的性能越好。
- 应用场景:在单细胞RNA测序数据的去噪和重构中,MSE用于评估去噪或重构数据与原始数据之间的差异。例如,在使用深度学习模型进行单细胞数据去噪时,MSE可以作为损失函数来指导模型的训练,通过最小化MSE来提高去噪效果。
决定系数(R²)
- 定义:衡量模型对数据变异性的解释程度。R²值越接近1,表示模型对数据的解释能力越强。
- 公式:$$R^2 = 1 - \frac{\sum _{i=1}^{n}(y_{i} - \hat{y}_{i})^2}{\sum _{i=1}^{n}(y_{i} - \bar{y})^2}$$ 其中,$y_{i}$ 是真实值,$\hat{y}_{i}$ 是预测值,$\bar{y}$ 是真实值的均值。
- 取值范围:R² 的取值范围在0到1之间。R² 越接近1,表示模型对数据的解释能力越强;R² 越接近0,表示模型对数据的解释能力越弱。
- 应用场景:在单细胞RNA测序数据的回归分析和预测建模中,R²用于评估模型的拟合优度。例如,在研究基因表达水平与细胞功能之间的关系时,可以使用R²来评估回归模型对基因表达变异的解释程度。
这些评估指标在单细胞RNA测序数据分析中,为研究人员提供了丰富的工具,帮助他们选择最佳的分析方法,提高研究的准确性和可靠性。
余弦相似度(CS)
- 用于衡量去噪数据与真实数据之间的相似性。CS值越高,表示两者越相似。
- 公式为:$$CS = \frac{\sum _{i=1}^{n} x_i y_i}{\sqrt{\sum _{i=1}^{n} x_i^2} \sqrt{\sum _{i=1}^{n} y_i^2}}$$ 其中,$x_{i}$ 和 $y_{i}$ 分别是两个向量的分量。
伪时间一致性评分(POS)
- 用于评估细胞轨迹推断的准确性。POS值越高,表示推断的伪时间顺序与真实顺序越一致。
- 公式为:$$POS = \frac{consistent_count}{consistent_count + inconsistent_count}$$ 其中,consistent_count 是一致的细胞对数,inconsistent_count 是不一致的细胞对数。
调整兰德指数(Adjusted Rand Index,ARI)
- 定义:用于衡量两个数据聚类结果之间的相似度。ARI值越高,表示聚类结果与参考标签越一致。
- 公式:$$ARI = \frac{\sum_{ij} \binom{n_{ij}}{2} - \left[\sum_i \binom{a_i}{2} \sum_j \binom{b_j}{2}\right] / \binom{n}{2}}{\frac{1}{2}\left[\sum_i \binom{a_i}{2} + \sum_j \binom{b_j}{2}\right] - \left[\sum_i \binom{a_i}{2} \sum_j \binom{b_j}{2}\right] / \binom{n}{2}}$$
其中:- $n_{ij}$ 是同时属于第 $i$ 个聚类和第 $j$ 个参考类别的对象数。
- $a_i$ 是第 $i$ 个聚类中的对象数。
- $b_j$ 是第 $j$ 个参考类别中的对象数。
- $n$ 是总对象数。
- 取值范围:ARI 的取值范围在 -1 到 1 之间。当两个聚类结果完全一致时,ARI 值为 1;当聚类结果与参考标签完全独立时,ARI 值接近 0;在某些情况下,如果聚类结果与参考标签的相似度低于预期,ARI 值可能会是负数。
- 应用场景:在单细胞RNA测序数据分析中,ARI常用于评估聚类算法的性能,通过将聚类结果与已知的细胞类型标签进行比较,来判断聚类结果的准确性。
标准化互信息(Normalized Mutual Information,NMI)
- 定义:用于衡量两个聚类结果之间的互依赖程度。NMI值越高,表示两个聚类结果之间的相关性越强。
- 公式:$$NMI = \frac{I(C; C’)}{\sqrt{H(C)H(C’)}}$$
其中:- $I(C; C’)$ 是聚类结果 $C$ 和参考聚类 $C’$ 之间的互信息。
- $H(C)$ 和 $H(C’)$ 分别是聚类结果 $C$ 和参考聚类 $C’$ 的熵。
- 取值范围:NMI 的取值范围在 0 到 1 之间。NMI 值为 1 表示两个聚类结果完全一致,值为 0 表示两个聚类结果完全独立。
- 应用场景:在单细胞RNA测序数据分析中,NMI常用于评估不同聚类算法之间的相似性,或者用于比较不同参数设置下的聚类结果。它可以帮助研究人员确定哪种聚类方法或参数组合能更好地识别细胞类型。
常用框架
scQCEA
- 是一个用于单细胞RNA测序数据质量控制和富集分析的R包,可以生成过程优化指标报告,用于比较样本集和可视化质量分数。
- 提供基于差异基因表达模式的自动化细胞类型注释。
scone
- 是一个灵活的框架,基于全面的数据驱动指标评估标准化方法的性能。
- 通过图形摘要和定量报告总结不同标准化方法的优缺点,并对大量标准化方法按性能进行排名。
SCORPIUS
- 用于重建单细胞的伪时间顺序,并计算伪时间一致性评分(POS)来评估细胞轨迹推断的准确性。
这些评估指标和框架为单细胞RNA测序数据的质量控制、标准化、去噪和聚类分析提供了全面的解决方案。
四、挑战与展望
技术挑战
- 数据稀疏性:scATAC-seq零值占比>90%,需定制化建模(如Binary PCA)。
- 跨模态对齐:RNA-ATAC非线性关联建模困难(如GLUE需知识图谱先验)。
- 计算可扩展性:百万级细胞分析内存优化(如Dask并行框架)。
前沿方向
- 深度学习:scGPT(通用单细胞大模型)支持跨模态预测。
- 动态建模:Waddington-OT整合时间序列与空间数据。
- 临床转化:肿瘤微环境靶点挖掘(如PD-1/PD-L1空间共定位)。
五、单细胞RNA测序数据分析与项目组织指南
1. 项目组织结构
为了确保单细胞RNA测序数据分析项目的高效管理,建议使用以下目录结构:
- Code 文件夹:存放代码,特别是需要运行的脚本(如
.py文件),这些不适合放在 Jupyter Notebook 中。 - Data 文件夹:存放所有
.h5ad文件,即数据分析过程中生成的文件。 - Figures 文件夹:存放在分析过程中生成的所有图形。
- Notebooks 文件夹:存放所有 Jupyter Notebooks 文件。
- Reports 文件夹:存放分析的报告、演示文稿或手稿草稿等文档。
- Tables 文件夹:存放表格文件,如不同ially expressed genes (DEGs) 之类的列表。
2. Jupyter Notebook组织结构
在Jupyter Notebook中,建议按以下步骤组织:
- Pre-processing:数据预处理步骤,包括加载数据、数据合并、质量控制、标准化、批次效应校正等。这部分完成后,可以将其保存为独立的 Notebook,以便日后回溯。
- Cluster Annotation:针对细胞群体的注释分析,即如何标记不同的细胞群体。
- RNA Velocity:分析RNA速度,类似于之前的课程中提到的分析方法。
- Pseudotime & Branching:伪时间与分支分析,分析细胞发展的时间轴和分支关系。
- Unsorted Requests:一般处理团队或合作伙伴的未分类请求。
可以根据需要添加其他模块,例如差异表达分析、基因集富集分析等。
3. 组织思路与小而精的Notebook
- 预处理阶段通常包含多个步骤,确保每个步骤都可以单独保存并回溯。完成预处理后,生成一个最终的
.h5ad数据对象并保存。 - 保持Notebook的简洁与小巧非常重要。大型Notebook容易变得缓慢,甚至会导致内存问题,超过100MB的文件可能无法保存。
4. 数据版本管理
- 建议保存多个版本的数据对象,以便不同的分析步骤能够访问不同版本的数据:
- Unfiltered version:未过滤的原始数据,通常在数据合并后保存。
- Normalized version:标准化后的数据。
- Batch Corrected version:批次效应校正后的数据。
- Annotated version:已注释的最终数据。
这些版本可以通过使用不同的“Layer”保存于同一对象中,便于回溯。
5. 可复现性(Reproducibility)
可复现性是当前生物数据分析中越来越重要的要求,尤其是对于代码分享与数据共享。
- 高影响力期刊通常要求共享代码,不再接受“按要求共享”的做法,因此确保代码与结果的可复现性非常关键。
- 在项目结束时,整理清理Notebook,确保其可以从头到尾运行而无错误。避免频繁跳跃运行不同的代码块,因为这可能导致结果不可复现并且引入错误。
6. 文档与版本控制
- 日期标签:为图形、表格等文件添加日期标签,可以帮助回溯和比较不同时间点的分析结果。
- 版本管理:随着分析的进行,往往会不断生成不同版本的图形和表格,明确记录每个版本的日期和修改内容有助于追溯与整理。
7. 环境管理
- 环境管理的重要性:当项目时间较长时(如半年或一年),环境(如Python版本、包版本等)可能会发生变化,导致不可复现的问题。
- 推荐使用虚拟环境:使用如Conda等工具创建并管理项目的虚拟环境。每个项目或分析步骤都可以拥有独立的环境,避免由于包更新导致结果的不同。
- 包版本固定:通过
conda或pip安装并锁定具体版本的包。 - Docker 容器:如果希望更加稳定地复现环境,可以使用Docker容器来固定包版本和操作系统环境,保证长时间后能够顺利回溯和复现。
- 包版本固定:通过
8. 结束时注意事项
- 在项目结束后,或者在回顾项目时,确保能够清楚地复现每个步骤。为此,要避免不必要的包更新或操作系统的更改。
- 每个操作都应详细记录,包括使用的包版本、Python版本等,以便日后查询和复现。
- 项目组织方式是个人化的,每个人可以根据自己的需求定制适合的结构。
- 从长远的角度考虑,设计一个清晰的结构,以便一年后能够轻松回顾、复制和复现以前的分析过程。